
Газета «Новости медицины и фармации» Кардиология (327) 2010 (тематический номер)
Вернуться к номеру
Эритропоэз и механизмы регуляции уровня железа плазмы крови
Авторы: В.И. Филимонов, Запорожский государственный медицинский университет
Версия для печати
Одной из проблем современной гематологии являются железодефицитные анемии, так как многие из них до сих пор плохо поддаются лечению. Нам представляется, что это вызвано неясностью некоторых механизмов их патогенеза, обусловленной недостаточной глубиной изученности процессов регуляции гомеостаза железа, его хранения в депо и доставки к костному мозгу — основному месту использования железа для синтеза гемоглобина.
Из 4–5 г железа, которое находится в организме, 20 % составля ет резервное железо, а остальное — функционально активное. Из этого количества в состав гемоглобина эритроцитов входит 62–70 %, 5–10 % содержится в скелетных мышцах (в миоглобине). Остальное железо находится в тканях, где оно принимает участие во многих метаболических процессах: в составе металлосодержащих энзимов обеспечивает митохондриальный транспорт электронов, синтез ДНК и деление клеток, метаболизм катехоламинов, детоксикационные механизмы, в обеспечении которых принимает участие, в частности, цитохром Р 450 , содержащий железо.
В обычных условиях для эритропоэза ежесуточно необходимо 20–25 мг железа. Практически все это железо костный мозг получает за счет его повторного использования. Основным местом резервного железа является ретикулоэндотелиальная система (РЭС) печени, откуда Fe поступает в эритробласты костного мозга с белком плазмы крови трансферрином (гликопротеином MB 76000), мигрирующим при электрофорезе белков плазмы вместе с b -глобулином. Поступившее в эритробласт железо используется для синтеза гема и депонируется в эритробласт в виде резерва. В макрофагах печени и костного мозга резервное железо депонируется в молекуле ферритина, в состав которой входит белок апоферритина, образующий подобие скорлупы, в центре которой аккумулируется железо. Молекулы ферритина, в свою очередь, образуют внутри лизосом большие аморфные нерастворимые агрегаты — гемосидерин. Таким образом, ферритин и гемосидерин — это формы резервного железа в клетках. При освобождении железа из клеточного резерва оно переводится в двухвалентное состояние (благодаря энзиму ксантиноксидазе, аскорбиновой кислоте и др.), соединяется с трансферрином и транспортируется через плазму крови к эритробластам костного мозга [1].
В настоящее время основным регулятором уровня железа в плазме крови большинство исследователей считает гепсидин (hepcidin). Гепсидин — небольшой протеин (25 аминокислот), который синтезируется в печени и здесь же на уровне купферовских клеток регулирует выход железа в русло крови. Впервые он был описан в 2001 году как один из стимуляторов антимикробного иммунитета [2, 3]. Но вскоре было показано и его участие в гомеостазе железа [4, 5].
Обстоятельно описаны также и механизмы его действия как одного из основных регуляторов гомеостаза железа на уровне обеспечения всасывания в энтероцитах тонкого кишечника [11], а также резорбции из клеток РЭС [11]. При поражении печени содержание гепсидина в плазме крови снижается, что приводит к дефициту усвоения железа из кишечника [8, 9].
Гепсидин стимулирует синтез РНК, что в конечном итоге связывает ферропортин и тем самым уменьшает поступление ионов железа в общий кровоток [11]. Одним из регуляторов уровня самого гепсидина является метриптаза-2 — фермент, расщепляющий гепсидин [12].
В то же время еще совсем неясным остается механизм, с помощью которого осуществляется взаимодействие системы эритропоэза (а это основной «потребитель» железа) и гомеостаза его в организме [6]. Лишь постулируeтся наличие какого-то еще неизвестного фактора-посредника [5, 7, 10]. Предполагается, что таким фактором может быть эритропоэтин [13].
Позволю себе напомнить некоторые положения современной науки об эритропоэзе и его основном стимуляторе — эритропоэтине, основным местом синтеза которого являются перитубулярные клетки почки [14], а также гепатоциты, окружающие венозные синусы печени [15]. В настоящее время при использовании различных методов культур тканей костного мозга показано, что эритропоэз осуществляется в так называемых эритробластических островках [16]. Превращение общей родоначальной клетки в эритроидные предшественники происходит при тесном межклеточном взаимодействии структур костного мозга и специфических регуляторов, а их дальнейшее созревание в зрелый эритроцит происходит при обязательном участии макрофагов костного мозга.
Эритроидный островок представляет собой центральный макрофаг, окруженный, как короной, эритробластами, находящимися на различной стадии своего созревания. Эти макрофаги обеспечивают модуляцию эритроидных колоний, продуцируя разнообразные специфические регуляторы активности влияния основного стимулятора пролиферации и созревания эритробластов — эритропоэтина [18]. Они же могут синтезировать некоторое количество эритропоэтина. В эритробластических островках развитие нормобластов заканчивается их денуклеацией (выталкиванием ядра) и превращением в ретикулоциты с последующим высвобождением этих клеток и поступлением их в синусоиды костного мозга. Ретикулоцит — это юный эритроцит, содержащий иРНК, благодаря которой еще в течение почти двух суток продолжается синтез гемоглобина: в первые сутки ретикулоцит находится в костном мозге, а во вторые — уже в русле крови. По их концентрации в периферической крови судят об интенсивности эритропоэза.
Главным стимулом продукции эритропоэтина является тканевая гипоксия, что подтверждается быстрым накоплением иРНК эритропоэтина после возникновения дефицита кислорода, к которому высоко чувствительны соответствующие клетки почек. Поэтому секреция эритропоэтина зависит от содержания кислорода в альвеолярном воздухе, функции легких (вентиляция, диффузия и перфузия), сердечного выброса, общей кислородной емкости крови, уровня гемоглобина, его способности связывать и отдавать кислород и определяется количеством кислорода в крови и потребностью тканей в нем [19, 20]. Между уровнями гемоглобина или гематокрита и плазменного эритропоэтина существует обратная линейная корреляция [20, 21]. Следовательно, уровень эритропоэтина в плазме может служить маркером тканевой оксигенации. По мнению некоторых исследователей, печень и почки выделяют неактивный эритропоэтин, так называемый эритроген, который в плазме крови под влиянием специфического фермента эритрогенина превращается в эритропоэтин. Эритропоэтин обладает рядом уникальных свойств [22–24]. Он не имеет сходства с каким-либо другим плазменным белком, а в плазме крови циркулирует только одна его форма. Плазменный клиренс эритропоэтина не зависит от его концентрации в плазме и от клеточности костного мозга. Исследователи не обнаружили преформированных мест его депонирования. Эритропоэтин оказывает влияние на различные звенья эритропоэза, начиная с реактивных клеток, позволяя им дифференцироваться в узнаваемые под микроскопом предшественники эритроцитов, в которых начинается синтез гемоглобина. Особенно активно он влияет на созревание эритроидных клеток-предшественников и, увеличивая их резерв, стимулирует пролиферацию созревающих клеток.
Суть исследования
Целью эксперимента явилось выяснение того факта, почему в норме при активации эритропоэза в плазме крови повышается концентрация железа; то есть является ли это ответом на уровень эритропоэтина плазмы, содержание в крови которого увеличивается, или нет. Указание на эритропоэтин как на стимулятор уровня железа в плазме крови возникает по аналогии с другими гормональными регуляторами. К примеру, гомеостаз йода, его всасывание стимулируется ТТГ, а концентрация кальция крови регулируется кальцитонином, паратгормоном и витамином D 3 .
Для этого нами использован многоступенчатый эксперимент. Сначала животных (крысы линии Вистар) на 18 часов помещали в барокамеру при пониженном содержании кислорода (аналог высоты 4000 м над уровнем моря). Это широко используемая модель стимуляции образования эритропоэтина. После этого крыс забивали кровопотерей, а сыворотку крови (по 2 мл) вводили реципиентам (№ 1). Спустя сутки, когда из их крови эритропоэтин уже исчезал [7], у этих животных также забирали кровь, а сыворотку вводили (по 2 мл) следующим реципиентам (№ 2). Спустя сутки кровь данных реципиентов исследовалась на содержание железа. В каждой серии также исследовалась концентрация железа у контрольных крыс (им вводился физиологический раствор).
Уровень железа и его производных в плазме крови исследовалися с помощью стандартного набора BIOTEST Fe70. Конечное определение экстинкции производилось на спектрофотометре на базе ЦНИЛа университета. Концентрация ретикулоцитов в крови определялась окраской мазков 1% спиртовым раствором бриллиант крезилблау.
Результаты и их обсуждение
Приведенная выше схема эксперимента позволила нам использовать два типа сыворотки: реципиентам № 1 вводилась сыворотка, содержащая эритропоэтин, а у реципиентов № 2 в сыворотке он уже отсутствовал, поскольку известно, что Т 1/2 вводимого с сывороткой эритропоэтин составляет всего 1,5 часа [7].
Полученные результаты исследований приведены в табл. 1. Прежде всего необходимо обратить внимание на то, что концентрация ретикулоцитов в крови животных отражает вышесказанное о наличии или отсутствии эритропоэтина во вводимой реципиентам сыворотке. У реципиентов № 1, так же как у животных после пребывания в гипоксической барокамере, как и должно быть под влиянием эритропоэтина, содержание ретикулоцитов возрастало. В отличие от этого у реципиентов № 2 оно оставалось без изменения, что свидетельствует об отсутствии у них повышенного уровня стимулятора.
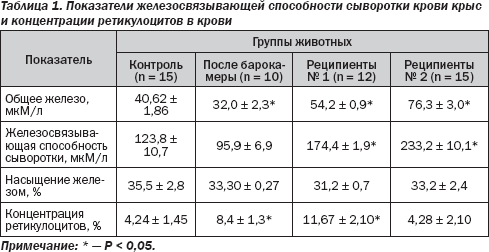
Как видно из приведенных данных, после пребывания в барокамере, которое приводит к стимуляции образования эритропоэтина и началу интенсификации кроветворения под его действием, уровень железа и железосвязывающая способность сыворотки крови достоверно понижались.
В отличие от этого введение сыворотки реципиентам № 1 приводит к росту уровня транспортного железа в их крови. Но наиболее важно то, что в еще большей степени возросли как уровень транспортируемого железа, так и железосвязывающая способность сыворотки крови у реципиентов № 2, причем отличие было достоверным даже по отношению к реципиентам № 1. Примечательно, что во всех группах животных процент насыщения железом транспортной системы плазмы был одинаковым.
Полученные результаты можно трактовать таким образом. Под воздействием стимулированного кроветворения у реципиентов № 1 увеличивается доставка железа к костному мозгу. Это обусловлено тем, что в их крови появляется какой-то фактор-посредник (его мы назвали фактором Fе), который информирует клетки-депо железа о необходимости выброса микроэлемента в кровь для обеспечения увеличенных потребностей костного мозга. Эффект этого посредника еще более четко проявляется у реципиентов № 2. Очень важно то, что он не является эритропоэтином, поскольку проявляет свой эффект и после исчезновения последнего.
Снижение показателей транспорта железа после пребывания в барокамере можно объяснить тем, что к моменту исследования данный фактор-посредник еще не образовался (или его еще очень мало), а активированное кроветворение уже забирает больше железа из плазмы крови.
Нельзя исключать того, что выявленный нами фактор является тем посредником, который информирует систему гепсидина об интенсивности кроветворения. Весьма важно то, что он не является эритропоэтином, вопреки мнению D.R. Asby et al. [ 13 ] . Косвенным подтверждением нашей точки зрения являются и результаты лечения недоношенных новорожденных с низким содержанием гемоглобина в эритроцитах, получавших эритропоэтин. При этом отмечена тенденция к снижению содержания сывороточного железа, ферритина, трансферрина даже при одновременном введении эритропоэтина и препаратов железа (от 2 до 6 мг/кг/сутки) и витамина Е (5– 15 мг/день) [25, 26]. Большинство исследователей отрицают преимущества повышенных лечебных доз железа для повышения эффективности терапии эритропоэтином и при других железодифицитных состояниях [27–29]. То есть можно предположить, что здесь имеет место недостаток образования фактора (фактора Fе), существование которого постулируется нами.
Выводы
1. При стимулированном эритропоэзе в плазме крови растет уровень железа и повышаются показатели железотранспортных систем плазмы крови.
2. Мобилизация железа из депо происходит под воздействием какого-то фактора, который мы обозначили рабочим названием «фактор Fe».
3. Фактор Fе не является стимулятором кроветворения эритропоэтином.
1. Руководство по гематологии / Под ред. А.И. Воробьева. — Т. 2. — М.: Медицина, 1985. — 366 с.
2. Krause А., Neitz S., Magert H.J., Sc hulz А., Forssmann WG., Schulz-Knappe P., Adermann К. LEAP-1, а novel highly disulfide-bonded human peptide, exhibits antimicrobial activity // FEBS Lett. — 2001. — 480. — 147-150.
3. Parc C.H., Valore E.V., Waring A.J., Ganz T. Hepcidin, а urinary antimicrobial peptide sinthsized in liver // J. Biol. Chem. — 2001. — 276. — 7806-7810.
4. Andrews N.C. Anemia of inflammation: the cytokine-hepcidin link // J. Clin. Invest. — 2004. — 113. — 1251-1253.
5. Peyssonnaux C., Zinkernagel A.S., Schuepbach R.A. Regulation of iron homeostasis by the hypoxia-inducible transcripcion factors (HIFs) // J. Clin. Invest. — 2007 Jul. — 117(7). — 1926-1932.
6. Wrighting D.M., Andrews N.C. Iron homeostasis and erythropoiesis // Curr. Top. Dev. Biol. — 2008. — 82. — 141-167.
7. Wieczorek L., Hirth P., Schope K.B. Molecular biology of Eryth ropoietin // Prod. Develop. Pharmac. — 1991. — Vol. 2. — Р. 13-16.
8. Toledano M., Kozer E., Goldstein L.H., Abu-Kishk I., Bar-Haim A., Siman-Tov Y., Rechavi M., Rechavi G., Weizer-Stern O., Berkovitch M. Hepcidin in acute iron tox icity // Am. J. Emerg. Med. — 2009. — 27(7). — 761-4.
9. Muckenthaler M.U. Fine tuning of hepcidin expression by positive and negative regulators // Cell Metab. — 2008. — 8(1). — 1-3.
10. Beguin Y. Soluble transferrin receptor for the evaluation of erythropoiesis and iron status // Clin. Chim. Acta. — 2003. — 329(1–2). — 9-22.
11. Ramey G. et al. Heptidin targets ferroportin for degradation in hepatocytes // Hematologica. — 2009 Sep. — 22. — 235-242.
12. Knutson M.D. Into the matrix: regulation of the iron regulatory hormone heptidin by metriptase-2 // Nutr. Rev. — 2009. —67(5). — 284-288.
13. Ashdy D.R. et al. Plasma heptidin levels are elevated but responsive to erythropoietin therapy in renal dis ease // Kidney Int. — 2009. — 75(9). — 976-81.
14. Lacombe C., Da Silva J.-L., Bruneval P. et al. Peritubular cells are the site of erythropoietin synthesis in the murine hypoxic kidney // J. Clin.Invers. — 1988. — Vol. 81. — P. 620-623.
15. Koury S.T., Bondarant M.C., Koury H.J. Localization of cells producing erythropoietin in murine liver by in situu hybridization // Blood. — 1991. — Vol. 77, № 11. — P. 2497-2506.
16. Bessis M. Ultrastructural aspects of erythropoiesis // International Conf. on Hematopoiesis. — Capri, 1971. — P. 7-20.
17. Захаров Ю.М., Тишевская Н.В. О взаимосвязи функциональной активности центральных макрофагов с кинетикой эритропоэза в эритробластических островках // Вопросы экспериментальной физиологии. — M.; Екатеринбург, 1997. — 95-103.
18. Захаров Ю.М., Рассохин А.Г. Эритробластический островок. — М.: Медицина, 2002. — 280 с.
19. Juul S.E. Erythropoietin in the neonate // Curr. Probl. Pediatr. — 1999. — Vol. 5. — P. 129-149.
20. Морщакова Е.Ф. Анемия недоношенных и эритропоэтин // Педиатрия. — 1997. — № 4. — С. 49-54.
21. Павлов А.Д., Морщакова Е.Ф., Румянцев А.Г. Эритропоэтин: биологические свойства и при менение в клинической практике. — М.: Гэотар Медицина, 2001.
22. Chen J. Anemia of premature // Amer. J. Perinatol. — 1995. — Vol. 5. — P. 314-318.
23. Ермоленко В.М., Николаев А.Ю. Эритропоэтин: биологические свойства и применение в клинической практике // Терапевтический архив. — 1991. — № 6. — С. 81-86.
24. Koenig J.M., Christensen R.D. Effect of erythropoietin on granulocytopoieiesis: in vitro and vivo studies in weanling rats // Pediatr. Res. — 1990. — Vol. 27. — P. 583-587.
25. Ohls R.K. The use of erythropoietin in neonates // Clin. Perinatol. — 2000. — Vol. 3. — P. 681-696.
26. Spencer M.K., Khong T.Y., Matthews B.L. et al. Haematopoietic indicators of fetal metabolic acidosis // Aust. NZJ Obstet. Gynaecol. — 2000. — Vol. 40. — P. 286-289.
27. Bader D., Kugelman A., Maor-Rogin N. et al. The role of high-dose oral iron supplementation during erythropoietin therapy for anemia of prematurity // J. Perinatol. — 2001. — Vol. 21. — P. 215-220.
28. Picaud J.C., Putet G., Salle B.L., Claris A.D. Iron supplementation in preterm infants treated with erythropoietin // Arch. Pediatr. — 1999. — Vol. 6. — P. 657-664.
29. Dani C., Reali M.F., Bertini G. et al. The role of blood transfusions and iron intake on retinopathy of pre maturity // Early Hum. Dev. — 2001. — Vol. 62. — P. 57-63.