
Международный неврологический журнал 4 (42) 2011
Вернуться к номеру
Симпозиум «Синдром мышечной гипотонии у новорожденных и детей раннего возраста»
Авторы: Евтушенко С.К., Морозова Т.М., Шаймурзин М.Р., Донецкий национальный медицинский университет им. М. Горького
Рубрики: Неврология
Версия для печати
Проводит: кафедра детской и общей неврологии ФИПО Донецкого национального медицинского университета им. М. Горького
Рекомендован: неврологам, педиатрам, неонатологам, семейным врачам
Синдром мышечной гипотонии (СМГ) у детей раннего возраста может представлять сложную диагностическую дилемму для клинициста. СМГ не имеет нозологической самостоятельности, входит в структуру различных синдромокомплексов и заболеваний, его основные клинические маркеры неспецифичны, а течение и исход вариабельны. Синонимами СМГ являются: «низкий мышечный тонус», «доброкачественная врожденная гипотония», «гипотоничный младенец», «врожденная мышечная гипотония», «врожденная мышечная слабость», «врожденная амиотония», «младенческая гипотония» и как крайний их вариант — «синдром вялого ребенка» (floppy baby syndrome). Данная терминология использовалась во многих случаях, когда не могла быть обнаружена какая-либо очевидная причина гипотонии. Однако эти определения не должны удовлетворять врача по той причине, что они объединяют все варианты мышечной гипотонии у детей независимо от этиологии и уровня нарушения регуляции мышечного тонуса.
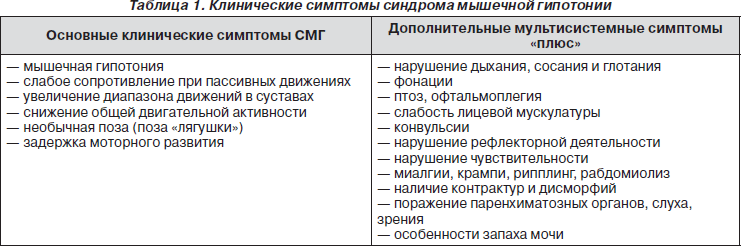
Диагностировать заболевания, в симптомокомплекс которых входит мышечная слабость и гипотония, помогают основные симптомы СМГ и их сочетание с дополнительными мультисистемными клиническими проявлениями (табл. 1). В настоящее время выделяют вариант изолированной мышечной гипотонии центрального или периферического происхождения и вариант СМГ с мультисистемными признаками — мышечную гипотонию «плюс».
Мышечная гипотония может быть обусловлена нарушением супрасегментарной регуляции альфа- и гамма-мотонейронов, гипофункцией спинальных альфа-мотонейронов, денервацией, повышением активности медиаторных ингибиторных механизмов, патологией ионных каналов, нарушением цитоскелета мышц и снижением энергообеспеченности мышечной ткани. Для сохранения физиологического мышечного тонуса и движений необходимо сбалансированное взаимодействие центральных нейрональных систем и морфологических элементов двигательной единицы. Гипотония выступает одним из основных симптомов их дисфункции и встречается при заболеваниях головного и спинного мозга, периферического нейрона, нарушениях нейромышечной передачи и патологии мышц. Однако центральные причины, проявляющиеся постуральной гипотонией, в структуре СМГ составляют 60–80 %, значительно превышая частоту возможных причин, связанных с двигательной единицей.
Выделяют следующие топические уровни происхождения СМГ:
1. Центральный уровень подразделяется:
— на церебральный уровень;
— спинальный уровень.
2. Периферический уровень (поражение двигательной единицы) подразделяется:
— на нейрональный и невральный уровень (болезни периферического нейрона);
— синаптический уровень (болезни нейромышечной трансмиссии);
— мышечный уровень (врожденные структурные миопатии, миодистрофии, миотонии, миоплегии, метаболические и воспалительные миопатии).
Топический уровень, вовлеченный в патологический процесс, можно определить на основании клинических данных (табл. 2).
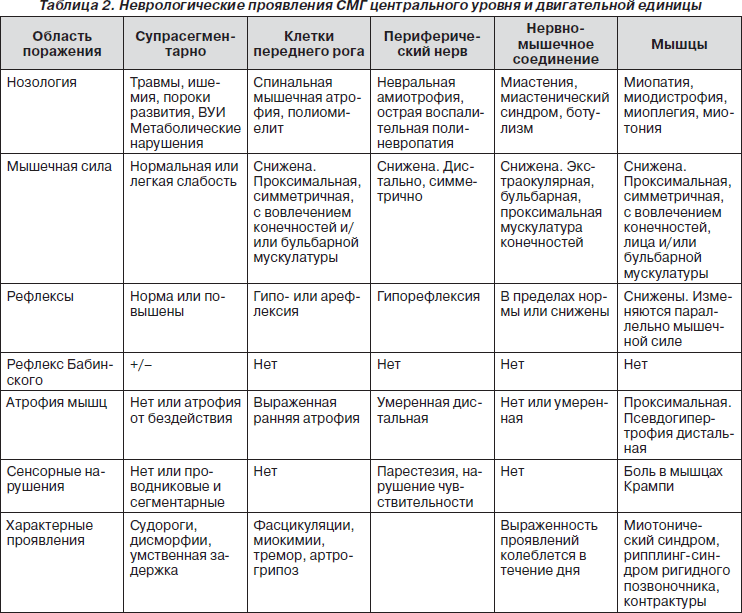
В случае центральной постуральной гипотонии мышечная сила сохранена, сухожильные рефлексы повышены, появляются патологические стопные знаки и клонусы, атрофия мышц не выражена. Церебральной гипотонии сопутствуют дополнительные симптомы: судороги, задержка редукции примитивных рефлексов и развития ребенка, дисморфические стигмы, вовлечение внутренних органов.
Поражение тела нижнего моторного нейрона (нейронопатия) подозревают у тех больных, у которых мышечная слабость сопровождается выраженной атрофией мышц и фасцикуляциями при отсутствии сенсорных нарушений. Сухожильные рефлексы чаще отсутствуют или резко угнетены. Показателями неврального процесса (невропатия) является слабость дистальной мускулатуры в сочетании с нарушением чувствительности и снижением рефлексов. О вовлечении синапса свидетельствует выраженная мышечная слабость поперечнополосатой, глазной и бульбарной мускулатуры. Особенно если степень мышечной слабости варьирует в течение суток, усиливается к вечеру и наблюдается феномен патологической утомляемости мышц. У большинства больных с миастенией рефлексы сохранены. Миопатия, в отличие от других заболеваний моторной единицы, обычно проявляется мышечной слабостью проксимальной локализации, объем пораженной мышцы не изменен или увеличен, рефлексы сохранены или незначительно снижены.
У детей с СМГ нарушается контроль поддержания позы, наблюдается невозможность ползания, астазия (неспособность стоять вследствие мышечной рассогласованности), деформация и переразгибание суставов, рекурвация в коленных суставах, вальгусные стопы.
Кроме того, у ребенка с гипотонией часто имеют место затруднения при кормлении, дыхательные проблемы, гастроэзофагеальный рефлюкс.
Однако верификация истинной причины и установление происхождения СМГ требуют вовлечения множества клинических методик и различных исследований. Опорным пунктом в установлении этиологии СМГ и индикатором выбора уместных диагностических тестов являются анамнестические и генеалогические данные, хронология процесса, оценка неврологического статуса. В ряде случаев клиницисты опираются не только на свои знания, но и на существующие возможности проведения дополнительных обследований. Поэтому всегда сохраняется вероятность, что окончательный диагноз не будет установлен.
Модифицированный алгоритм пошаговой диагностики СМГ у младенцев приведен в табл. 3.
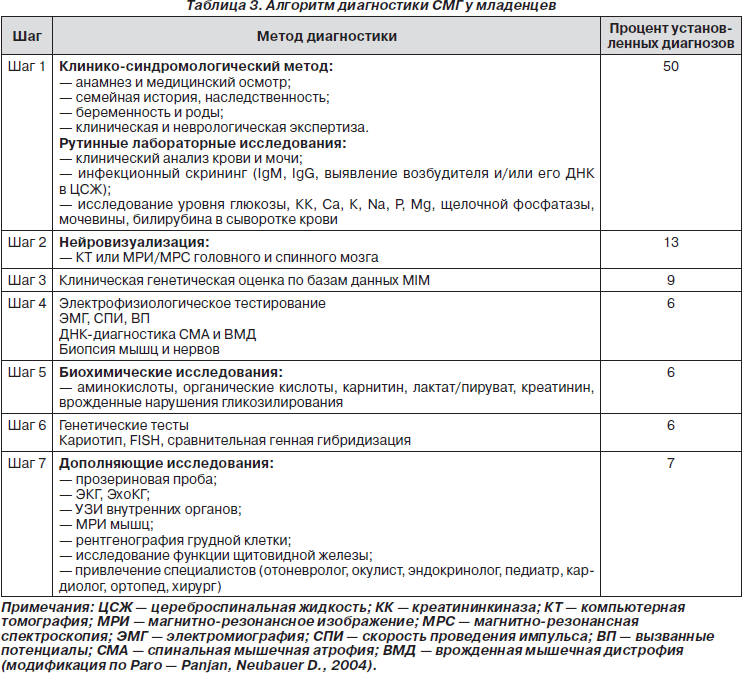
Клинико-синдромологический метод диагностики
Клинико-синдромологический метод диагностики базируется на изучении истории болезни, осмотре, выявлении пороков и дисморфий, определении локализации поражения на основе данных неврологического статуса. При этом важно выяснить, является ли мышечная слабость генерализованной (диффузной) или локальной, каково начало (острое или постепенное), наличие провоцирующих факторов и характер течения мышечной слабости (эпизодический, интермиттирующий, рецидивирующий или прогрессирующий). Особенности и сроки возникновения мышечной гипотонии и последующего ее течения служат важными факторами в диагностических алгоритмах.
Первый шаг в оценке младенца с гипотонией — это изучение семейного анамнеза, пренатальных и перинатальных факторов. Пренатальный анамнез должен включать информацию относительно инфекционных, токсических или наследственных заболеваний у матери, эмбрионального развития и внутриутробной активности плода. Слабые движения плода или их отсутствие могут свидетельствовать о нейрональном процессе (спинальной мышечной амиотрофии). Интранатальная история может помочь идентифицировать травматический или инфекционный фактор, приводящий к СМГ.
Новорожденного с низкой оценкой по шкале Apgar, вялостью и гипотонией нужно рассматривать как угрожаемого по септическому состоянию, пока не будет доказан другой генез. Асфиксия, краниоцервикальная и спинальные травмы, ассоциированные с акушерскими пособиями в родах, патологическим предлежанием плода, будут манифестировать гипотонией и другими признаками критического состояния или спинального шока. Неврологические симптомы могут сохраняться дни и недели. После острого периода церебральный или спинальный уровень гипотонии становится более очевидным, появляется дистония, повышаются сухожильные рефлексы или развиваются парезы спинального типа.
У младенца, рожденного в срок здоровым, но у которого через 12–24 часа развивается вялость, гипотония и признаки нейродистресс-синдрома, возможно, имеет место врожденное нарушение метаболизма.
Транзиторная миастения новорожденного с высокой вероятностью будет у ребенка, рожденного от матери с миастенией. Диагноз миотонической дистрофии может быть установлен в случае дистонии матки в родах, сложностей потужного периода и выявления миотонического феномена у матери (например, при крепком рукопожатии мать не может быстро расслабить кисть).
Осмотр новорожденного и младенца. Клиническая оценка включает детальный неврологический осмотр, исследование тонуса, силы и рефлексов. При исследовании мышечного тонуса у младенца необходимо обращать внимание на консистенцию мышц, определяемую пальпаторно, и характер пассивных движений (амплитуда и мышечное сопротивление при движении конечности). Осматривая ребенка, необходимо исследовать как пассивный, так и активный мышечный тонус и не просто фиксировать их изменение, а отмечать изменения в отдельных мышечных группах.
Важно отличать мышечную слабость от гипотонии. Гипотония трактуется как снижение сопротивления с гипермобильностью в суставах при пассивных движениях в сочетании с ухудшением способности поддерживать антигравитационный постуральный двигательный контроль. Слабость — это снижение мышечной силы. Вялые младенцы всегда имеют мышечную гипотонию, но гипотония может существовать без мышечной слабости. Наиболее широко миологами используется шкала определения степени мышечной слабости (по R.D. Adams и др., 2005 г.):
0 — паралич;
1 — минимальные сокращения;
2 — активное движение при устранении силы тяжести;
3 — слабое сокращение против силы тяжести;
4 — активное движение против силы тяжести и сопротивления;
5 — нормальная сила.
У здорового доношенного ребенка отмечается так называемая эмбриональная поза. Вследствие флексорного повышения тонуса руки согнуты во всех суставах, приведены к туловищу и прижаты к грудной клетке, кисти сжаты в кулаки, большие пальцы кистей рук лежат под четырьмя остальными; ноги согнуты в суставах и отведены в бедрах, в стопах преобладает тыльное сгибание. В ряде случаев отмечается преимущественное физиологическое повышение мышечного тонуса в руках по сравнению с ногами и экстензия шеи. Мышечный тонус может меняться в зависимости от физиологического состояния ребенка. Поэтому нестойкие и незначительные вариации не надо рассматривать как заведомо патологические. Меняющийся тонус в одной и той же группе мышц называется мышечной дистонией.
Снижение мышечного тонуса может быть локальным и генерализованным (диффузным). При генерализованном снижении мышечного тонуса отмечается специфическая поза новорожденного — поза «лягушки» (разогнутые во всех суставах конечности, бедра отведены и наружно ротированы, живот широкий и уплощенный). Диапазон пассивных движений увеличен, при подвешивании лицом вниз голова и конечности свисают, при тракции отсутствует фаза сгибания и голова запрокидывается назад. Диффузная мышечная гипотония может быть признаком большинства соматических и неврологических заболеваний периода новорожденности (сепсис, пневмония, респираторный дистресс-синдром, внутриутробные инфекции различной этиологии, метаболические нарушения, II–III стадия гипоксически-ишемической энцефалопатии, внутричерепные кровоизлияния, спинальная родовая травма, нервно-мышечные заболевания). Локальная гипотония может быть обусловлена невральными (травматическая невропатия, плексопатия) или сегментарными (спинальная травма) нарушениями, соответствующими иннервации.
Определение пассивного мышечного тонуса основано на ощущении сопротивления при пассивном сгибании и разгибании конечностей. Имеет значение амплитуда пассивных движений, которая измеряется величиной угла между мобильным дистальным и фиксированным проксимальным отделом конечности или туловища. Величина угла дает представление о степени растяжения определенных групп и отдельных мышц (предплечья, плеча, кисти, тазового пояса, шеи и др.). Ориентировочными показателями нормального пассивного мышечного тонуса являются следующие параметры: при повороте головы подбородок касается акромиального отростка; разгибание рук в локтевых суставах возможно до 180°; сгибание в лучезапястных суставах — до 150°; отведение в сторону согнутых бедер — на 75° в каждую сторону; разгибание ноги в коленном суставе при согнутом под углом 90° бедре возможно до 150°; дорсальное сгибание стоп составляет 120°.
Активный мышечный тонус. Показателями состояния активного мышечного тонуса являются поза ребенка, скорость возвращения разогнутой конечности в исходное положение, сила движений, что позволяет судить о статической и фазовой мышечной активности. Поза ребенка характеризует статическую мышечную активность, а тонус мышц во время движения — фазовую. В положении на спине все младенцы с СМГ выглядят одинаково независимо от причины и локализации поражения. Спонтанные движения ограничены или отсутствуют. В противоположность нормальной позе бедра полностью отведены кнаружи, руки вытянуты вдоль туловища или находятся в состоянии незначительного сгибания. Вследствие слабости мышц грудной клетки формируется ее воронкообразная деформация. У младенцев, лежащих неподвижно, может развиваться уплощение затылка. Когда ребенка сажают, его голова падает вперед, плечи опускаются, конечности свисают. У новорожденных с внутриутробной гипотонией уже при рождении могут диагностироваться дислокация тазобедренных суставов, артрогрипоз или их сочетание. Дислокация тазобедренных суставов — частое проявление внутриутробной гипотонии, поскольку для нормальной фиксации головки бедра в ацетабулярной впадине требуется полноценное сокращение мышц. Артрогрипоз варьирует по тяжести от наиболее распространенной косолапости до симметричных сгибательных деформаций.
Тонус у младенцев с симптомами гипотонии может быть оценен при проведении пробы на тракцию, проб с вертикальным и горизонтальным подвешиванием.
Проба на тракцию — наиболее чувствительный тест для определения постурального тонуса. Врач захватывает кисти ребенка и подтягивает его в положение сидя. У здорового ребенка голова приподнимается от поверхности сразу вслед за телом. Когда достигается сидячее положение, голова удерживается прямо посередине. Во время пробы исследователь ощущает сопротивление тракции и наблюдает сгибание конечностей ребенка в локтевых и коленных суставах. У доношенных детей допустимо лишь минимальное запаздывание движения головы. Аномальная проба на тракцию в виде большой задержки движения головы вслед за туловищем и отсутствие сгибания в конечностях у доношенного новорожденного указывают на гипотонию. Проба на тракцию не может быть проведена у недоношенных, не достигших 33 недель гестации. Возможности сгибателей шеи поднять голову появляются после 33 недель.
Вертикальное подвешивание. Врач располагает свои кисти в подмышечных областях ребенка и поднимает его вверх. Пока ребенок находится в вертикальном положении, его голова располагается прямо по средней линии, а конечности сохраняют позу сгибания в тазобедренных, коленных и голеностопных суставах. У ребенка с мышечной гипотонией при вертикальном подвешивании голова свисает вперед. Ноги свободно свисают. Из-за слабости мышц плечевого пояса ребенок не удерживается и «проскальзывает» между руками исследователя. «Проваливание» головы ребенка и симптом «вялых плеч» у доношенных новорожденных возникает как при гипоксических парасагиттальных поражениях головного мозга, так и при травме шейного отдела спинного мозга.
Горизонтальное подвешивание. Здоровый младенец поднимает голову, удерживает спину прямо, сгибает конечности в локтевых, тазобедренных, коленных и голеностопных суставах. Может периодически разгибать голову, напрягать спину и группироваться, преодолевая силы гравитации. Гипотоничный новорожденный и младенец перегибаются через руки врача как перевернутая буква «U», при этом голова и ноги свободно свисают (симптомы «тряпичной куклы» или «провисшего белья»).
Оценка массы мышц. Выявить атрофию или гипертрофию мышц у детей младшего возраста часто довольно трудно из-за большой вариабельности этого показателя у здоровых детей. Атрофию мышцы легче установить при наличии асимметрии. Следует помнить о так называемой псевдогипертрофии мышц. Это бывает достаточно выражено при некоторых мышечных дистрофиях и спинальной мышечной атрофии, когда мышечная ткань замещается соединительной или жировой тканью.
При осмотре ребенка необходимо обратить внимание на наличие фасцикуляций, миокимий и других проявлений спонтанной мышечной активности, которые свидетельствуют о вовлечении мотонейрона.
Некоторые болезни, такие как миотоническая дистрофия и лицеплечелопаточная дистрофия, имеют патогномоничный лицевой фенотип. Менее характерными, но достаточно очевидными признаками слабости лицевой мускулатуры отличаются другие миопатии и миастения.
Наличие ретракции ахиллова сухожилия, контрактур голеностопных и тазобедренных суставов, а также сколиоза свидетельствует о том, что мышечная слабость существует уже долгое время.
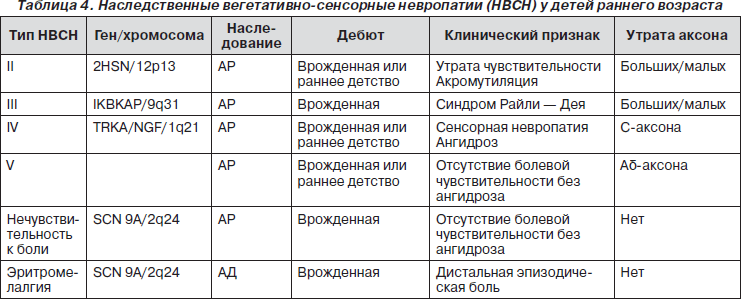
Сенсорное тестирование. Симптомы чувствительных расстройств обычно предполагают заболевание периферической нервной системы, однако так же, как и слабость, расстройства чувствительности могут сопровождать неврологические поражения на любом уровне нервной системы. В раннем детском возрасте дебютируют следующие вегетативно-сенсорные невропатии (табл. 4).
Поражение сердца. Большинство заболеваний скелетной мускулатуры, как правило, сопровождается изменениями в сердечной мышце. Довольно специфические электрокардиографические изменения возникают при дистрофии Дюшенна и дефиците кислой мальтазы. У больных с миотонической дистрофией, митохондриальными заболеваниями, каналопатиями могут возникнуть нарушения сердечной проводимости, включая полную поперечную блокаду. Во всяком случае, ЭКГ необходимо сделать всем больным с нервно-мышечной патологией, особенно больным с миопатиями.
Патология системы органов дыхания. Ослабление функции легких у больных с острыми и хроническими нервно-мышечными заболеваниями может прогрессировать до дыхательной недостаточности. Ранним проявлением ослабления дыхательной мускулатуры является снижение максимального экспираторного и инспираторного давления. Особенно значительно у больных с нервно-мышечными заболеваниями выражена слабость диафрагмы. У больных со слабостью диафрагмы функциональные легочные нарушения более отчетливо проявляются в горизонтальном положении, у них отмечаются также парадоксальные движения брюшной стенки. Больные с хронической дыхательной недостаточностью даже в домашних условиях нуждаются в поддержании дыхания.
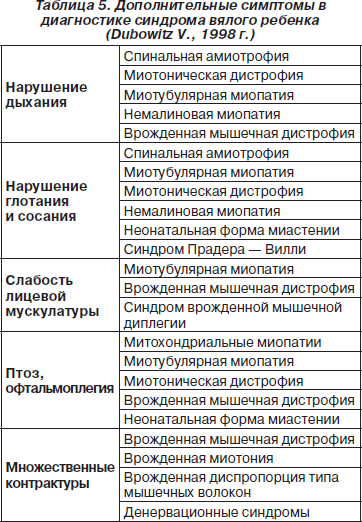
Дифференциальная диагностика синдрома вялого ребенка по дополнительным симптомам представлена в табл. 5.
Острая генерализованная мышечная слабость. Общая мышечная слабость, развивающаяся в течение менее чем одного часа, обычно бывает связана с метаболическим или токсическим поражением синапса или самой мышцы. Изменения концентрации калия, кальция, натрия, магния или фосфора в крови могут обусловить частичный или полный паралич мышцы. Так, острая недостаточность нервно-мышечного соединения с нарушением его трансмиссивной функции возникает при ботулизме, гипермагнезиемии, при применении аминогликозидных антибиотиков, фенитоина, высоких доз кортикостероидов и некоторых других препаратов. Мышечная слабость, развивающаяся в течение суток, может сопровождать метаболические нарушения, изменения электролитного баланса, интоксикацию, периодический паралич, острые воспалительные миопатии, особенно вызываемые вирусами или паразитами, а также некоторые острые воспалительные полиневропатии (синдром Ландри).
Подострая мышечная слабость. Мышечная слабость, развивающаяся в течение нескольких дней, более характерна для поражения периферического нерва или нервно-мышечного соединения, чем для поражения мышц или клеток передних рогов. Острая воспалительная полирадикулоневропатия (синдром Гийена — Барре), а также токсические невропатии — порфирийная, дифтерийная и другие — характеризуются подострым началом. При дифференциальной диагностике нужно учитывать миастению и другие нарушения синаптической трансмиссии. Подострая мышечная слабость может возникнуть при полимиозите, дерматомиозите, эндокринной патологии.
Слабость бульбарных мышц (ринолалия, покашливание, нарушение кашлевого рефлекса) возникает обычно в случае поражения моторного нейрона, синапса (миастения) и мышц (структурная миопатия, окулофарингеальная и миотоническая дистрофия). Слабость глазных мышц и птоз характерны для миастении, но не характерны для поражения моторного нейрона и периферической нейропатии. Офтальмопарез типичен для миастении, но может возникнуть также при миотонической и окулофарингеальной дистрофии. Офтальмоплегия характерна для митохондриальных заболеваний, таких как синдром Keams — Sayre.
Особое внимание необходимо уделить оценке неврологического статуса недоношенного новорожденного, у которого гипотония мышц является этапом развития и становления моторики.
28-недельный недоношенный младенец очень вялый. Если положить его животом на ладонь, он будет свисать без попытки к разгибанию шеи, конечностей и спины.
34-недельный младенец удерживает ноги в положении «ног лягушки», тогда как в руках отмечается гипотония и разгибание.
К 38-й неделе должно отмечаться сгибание всех конечностей.
К 34-й неделе появляется некоторая реакция опоры, а к 38-й неделе эта реакция уже хорошо развита.
До 30 недель голова совершенно не удерживается. К 38-й неделе отмечается разгибание шеи вслед за туловищем, хотя в вертикальном положении голова не удерживается.
Поисковый рефлекс до 30 недель не вызывается. На 30-й неделе слаб и имеет большой латентный период. К 34-й неделе этот рефлекс полностью развит.
Сосательный рефлекс формируется к 34-й неделе. До этого времени рефлекс слабый, с длительным латентным периодом, сосательные движения вызываются, но координация сосания, глотания, дыхания может быть несовершенной.
Ладонно-ротовой рефлекс Бабкина вызывается с 28-й недели.
Назопальпебральный рефлекс отсутствует до 32-й недели, полностью развит после 34 недель.
Рефлекс Робинсона до 31–32-й недели проявляется сгибанием пальцев, с 32-й недели — сжиманием пальцев в кулак, а с 36-й — полностью выражен. Нижний хватательный рефлекс Веркома вызывается с 28-й недели и с возрастом становится более выраженным.
Рефлекс Моро на 28-й неделе развит частично, фаза приведения отсутствует. К 32-й неделе появляется приведение верхних конечностей, а к 38-й неделе может быть вызван в полном объеме.
Перекрестный разгибательный рефлекс (при раздражении подошвы одной ноги, находящейся в положении разгибания, другая нога сгибается и отдергивается, а затем выпрямляется с разведением пальцев) до 36-й недели отсутствует.
Шаговый рефлекс у 28-недельных младенцев отсутствует. В возрасте 32–34 недель появляется в виде ходьбы с опорой на пальцы стопы. Доношенный младенец ступает на всю стопу.
Защитный рефлекс у недоношенных вызывается через 3–5 секунд после латентного периода.
Однако здоровый недоношенный младенец к концу третьего триместра внеутробной жизни должен соответствовать уровню статомоторного и когнитивного развития ребенка, рожденного в срок!
Синдром мышечной гипотонии центрального уровня
Для церебрального уровня СМГ клинико-электронейромиографическими маркерами являются: церебральное угнетение или возбуждение, асимметричность мышечной гипотонии, сохранность мышечной силы, нормо- или гиперрефлексия, клонусы, патологические рефлексы и позы, задержка редукции примитивных рефлексов и становления постуральных рефлексов; перекрест ног при вертикальном подвешивании, конвульсии, респираторный дистресс, поражение паренхиматозных органов. Но наиболее важно наличие таких церебральных нарушений, как угнетение сознания и судорожные пароксизмы.
У младенцев с дисморфическими чертами и пороками внутренних органов гипотония обычно детерминирована аномалиями развития головного мозга. О наличии спастичности, указывающей на церебральную патологию, можно судить по характерной «кулачковой» установке кистей, перекресте бедер при вертикальном подвешивании. Позотонические рефлексы у новорожденных и грудных детей с церебральной гипотонией выявляются даже в тех случаях, когда их спонтанные движения ограничены. Рефлекс Моро может быть очень ярким в случае острой энцефалопатии, особенно метаболического происхождения. Важным маркером церебральной патологии является наличие выраженных или постоянных тонических шейных рефлексов, а также их сохранение после 4 мес. жизни. Сухожильные рефлексы преимущественно в норме, но могут быть повышены и сопровождаются клонусом.
ЭМГ — «залповые» группировки осцилляций, укорочение латенций М-ответа, Н-рефлекса, повышение амплитуды и площади М-ответа и увеличение СПИ по двигательным волокнам.
Основные причины церебральной мышечной гипотонии:
— гипоксически-ишемическая энцефалопатия;
— родовая травма; внутричерепное кровоизлияние;
— перинатальный дистресс;
— внутриутробные инфекции;
— постнатальные заболевания.
Хромосомные болезни:
— синдром Прадера — Вилли (15q 11–13);
— трисомии;
— другие генетические дефекты;
— церебральная мальформация.
Метаболические и мультисистемные заболевания:
— дефицит кислой мальтазы;
— дефицит цитохром-С-оксидазы;
— неонатальный дефицит фосфофруктокиназы;
— неонатальный дефицит фосфорилаз;
— гликогеноз;
— ганглиозидоз;
— пероксисомные болезни;
— цереброгепаторенальный синдром;
— митохондриальные энцефаломиопатии;
— первичный дефицит карнитина;
— окулоцереброренальный синдром (Лоу-синдром).
Стандарт диагностики СМГ церебрального уровня:
— КТ, МР-визуализация, МР-спектроскопия, НСГ;
— ЭЭГ, ВП;
— ЭКГ, ЭхоКГ, УЗИ внутренних органов;
— биохимические исследования;
— инфекционный скрининг (ЦСЖ, кровь);
— метаболический скрининг;
— генетическое тестирование;
— консультация других специалистов.
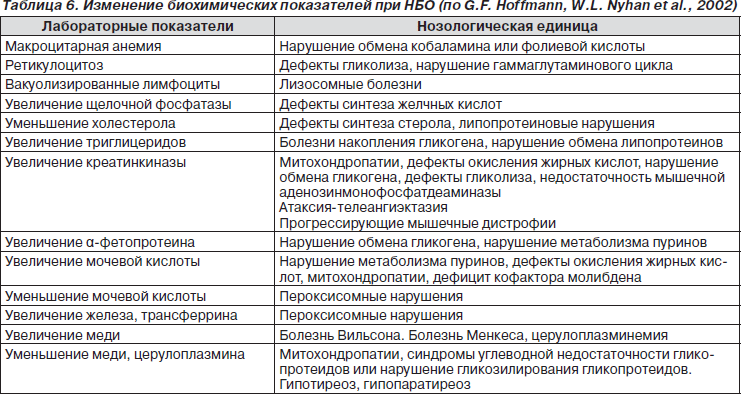
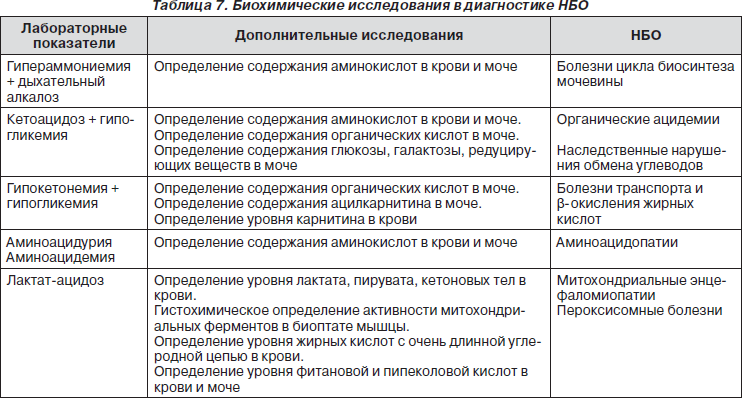
В табл. 6 и 7 показана высокая информативность традиционных биохимических исследований, представленных с позиций наследственных болезней обмена (НБО).
Синдром мышечной гипотонии спинального уровня
Маркерами СМГ спинального уровня являются: гипо-и атония, гипо- и арефлексия в фазе спинального шока. В дальнейшем наличие парезов спинального типа, сочетание проводниковых и сегментарных нарушений чувствительности и функции тазовых органов, дыхательные расстройства. На уровне скрытых и явных дисморфий часто располагаются: spina bifida, спинальный дермальный свищ, нейроэктодермальные отростки, атрофические изменения, локальный гипертрихоз, факосы.
Основные причины спинальной мышечной гипотонии:
— спинальная травма;
— ишемическая миелопатия (травматическая, септическая);
— аномалия спинного мозга;
— опухоль спинного мозга.
Стандарт диагностики СМГ спинального уровня:
— КТ, МР-визуализация;
— рентгенография позвоночника;
— ЭНМГ, соматосенсорные вызванные потенциалы (ССВП).
Радиологические исследования визуализируют: кровоизлияние, отек, разрыв спинного мозга, нарушение целостности, подвывихи и вывихи позвонков в случае травмы. В случае аномалий спинного мозга покажут миеломенингоцеле, фиксированный спинной мозг, сирингомиелию, нейроэнтеральные кисты, сосудистые мальформации, диастематомиелию, расщепление дужек позвонков.
Болезни двигательной единицы представлены следующими уровнями и основными нозологическими группами:
1. Периферический мотонейрон:
— спинальные мышечные атрофии (СМА);
— невральные мышечные атрофии.
2. Синаптический уровень:
— миастения;
— миастенические синдромы;
— ботулизм.
3. Мышечный уровень:
— структурные миопатии;
— миодистрофии;
— миотонии;
— миоплегии.
Стандарт диагностики болезней двигательной единицы
— КК, креатинин, Са, К, Na, Р, Mg;
— ДНК- тесты, прозериновая проба;
— электродиагностика (ЭНМГ, ССВП);
— ЭКГ;
— МРИ мышц;
— мышечная и невральная биопсия;
— консультация окулиста, отоневролога, ортопеда.
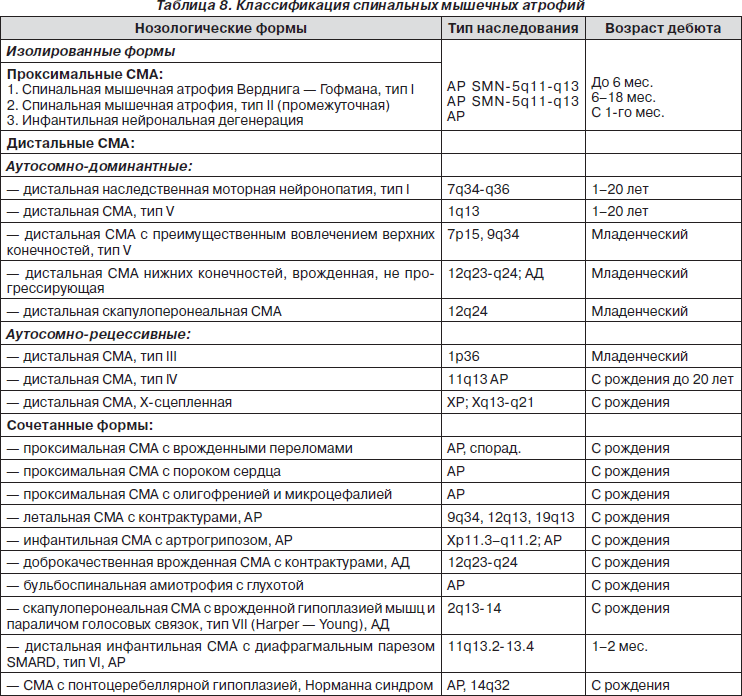
СМГ нейронального уровня (спинальные мышечные атрофии). Болезни периферического двигательного нейрона представляют собой гетерогенную группу прогрессирующих заболеваний генетической природы, наследуемых чаще по аутосомно-рецессивному (АР) или Х-сцепленному типу (табл. 8). Дегенерация мотонейронов в спинном мозге, двигательных ядрах ствола мозга приводит к возникновению симметричного вялого паралича поперечнополосатых мышц конечностей и туловища. Большинство описанных клинических вариантов СМА являются изолированными, то есть симптомы вялого паралича или пареза являются единственным клиническим проявлением болезни. В группу сочетанных форм СМА можно включить ранние детские варианты с микроцефалией, умственной отсталостью, врожденными переломами, артрогрипозом, пороками сердца. При морфологическом исследовании мышц выявляются специфические признаки мышечного поражения в виде избыточной неравномерности диаметра мышечных волокон: скопления уменьшенных в размере волокон («пучковая» атрофия) чередуются с участками гипертрофированных волокон. Наибольшую распространенность имеют проксимальные формы с аутосомно-рецессивным типом наследования, которые составляют 80–85 % от всех наследственных СМА. Следующей по частоте встречаемости является группа дистальных спинальных амиотрофий, нозологические формы которых составляют не менее 10 % всех СМА.
Учитывая, что распространение мышечной слабости при дистальных СМА сходно с таковым при поражении периферических нервов, ряд авторов обозначает дистальные СМА как дистальные наследственные моторные нейронопатии (dHMN).
Отличительными особенностями дистальных СМА по сравнению с наследственными моторно-сенсорными нейропатиями (НМСН) являются: отсутствие чувствительных нарушений, длительная сохранность сухожильных рефлексов, нормальная скорость проведения импульса по периферическим нервам и отсутствие изменения сенсорного потенциала.
Клинико-электронейромиографические маркеры СМА: симметричный характер вялого пареза с доминированием в проксимальных мышечных группах, арефлексия и атония, фасцикуляции. Отсутствие чувствительных нарушений.
На ЭНМГ выявляются характерные признаки денервации вследствие поражения мотонейронов — спонтанная ритмическая активность («ритм частокола»), потенциалы фибрилляций, потенциалы фасцикуляций, положительные острые волны, изменение ПДЕ с формированием гигантских полифазных потенциалов, уменьшение числа двигательных единиц.
Классификация СМА представлена в табл. 8. Подтверждение формы СМА проводится с помощью генетического обследования и выявления мутантного гена.
Дифференциальная диагностика инфантильных и ранних детских форм СМА включает:
— врожденные мотосенсорные невропатии;
— врожденные гипомиелинизирующие невропатии;
— неонатальную и врожденную миастению;
— ботулизм;
— синдром Дауна;
— синдром Марфана;
— синдром Прадера — Вилли;
— метаболические болезни (органические ацидурии и митохондриальные болезни);
— дефицит кислой мальтазы (гликогеноз II типа);
— адренолейкодистрофию;
— болезнь Гоше;
— синдром Хурлера;
— GM1 ганглиозидоз;
— поражение спинного мозга;
— полиомиелит;
— острую воспалительную полиневропатию.
СМГ неврального уровня (невральные мышечные атрофии). Наследственные моторно-сенсорные нейропатии (НМСН) у младенцев — группа генетически гетерогенных заболеваний с прогрессирующим поражением периферических нервов (табл. 9).
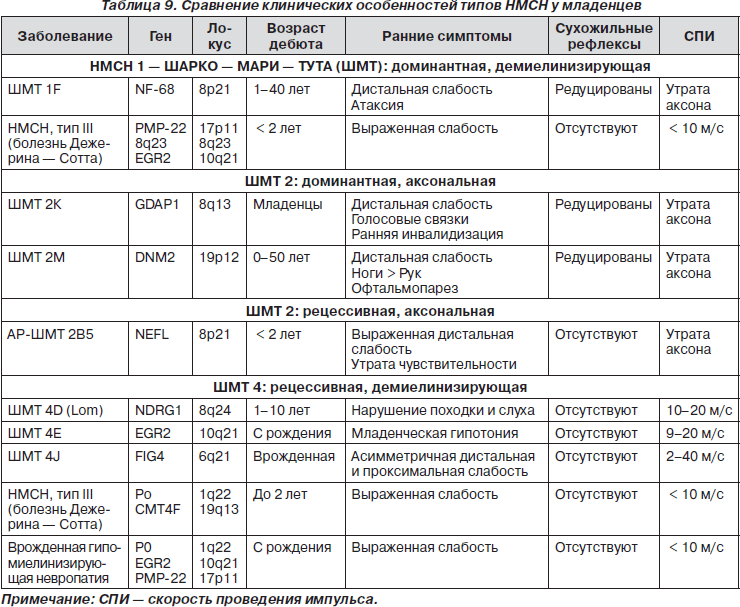
Клинические проявления характеризуются сочетанием слабости и атрофии мышц дистальных отделов рук и ног с последующей их деформацией, расстройствами чувствительности, трофическими и вегетативными нарушениями, сухожильной гипо- или арефлексией. Достаточно часто к указанным симптомам поражения периферических нервов присоединяются расстройства координации, зрения, слуха, эндокринные нарушения. Клиническая картина значительного числа генетических вариантов НМСН имеет выраженное сходство, что затрудняет их дифференциацию на клиническом уровне.
Основными ЭНМГ-признаками являются: 1) резкое снижение скоростей проведения импульса по периферическим нервам, которое в среднем составляет 17–20 м/с и колеблется от 5 до 34 м/с; 2) снижение амплитуды М-ответа; 3) удлинение дистальной латенции и F-волны; 4) отсутствие или резкое снижение амплитуды сенсорного потенциала.
В биоптате периферических нервов определяются специфические луковицеподобные утолщения миелиновой оболочки периферических нервов, образованные отростками шванновских клеток и базальной мембраны, чередующиеся с участками де- и ремиелинизации.
СМГ синаптического уровня
Миастения (myastenia gravis) — это патологическое состояние, связанное с нарушением иммунных механизмов, которые влияют на нервно-мышечную передачу, и характеризующееся феноменом патологической мышечной утомляемости. В основе патогенеза приобретенной миастении лежит аутоагрессия против ацетилхолиновых рецепторов (AChR) постсинаптической мембраны поперечнополосатых мышц.
Неонатальная миастения — преходящее состояние, наблюдающееся у младенцев, родившихся от матерей, страдающих миастенией, и обусловленное переходом через плаценту материнских антител к ацетилхолиновым рецепторам. Первые симптомы заболевания плода могут проявляться снижением его двигательной активности. У новорожденных манифестация первых признаков происходит через час после рождения. Мышечная гипотония и общая слабость выступают на первый план в клинической картине. Отмечается маскообразное лицо — гипомимия, опущение век — птоз, тихий голос, слабое сосание, глотание. Постепенно развивается бульбарный синдром. Диагноз подтверждается проведением пробы с прозерином подкожно 0,15 мг/кг, после которой наступает улучшение общего состояния, уменьшаются дыхательные расстройства, появляется крик, повышается мышечный тонус и восстанавливается рефлекс Моро. По данным ЭМГ подтверждено, что транзиторная форма неонатальной миастении может продолжаться в течение 6–12 недель. В терапии используется прозерин и плазмаферез.
Большинство младенцев, рожденных от матерей с миастенией, обладают анти-AChR антителами при рождении. Высокие материнские серологические уровни антител к AChR до рождения можно снизить, назначив плазмаферез, что предотвратит проявление миастении в неонатальном периоде.
Формы myastenia gravis (МG) в неонатальном и детском возрасте представлены в табл. 10.
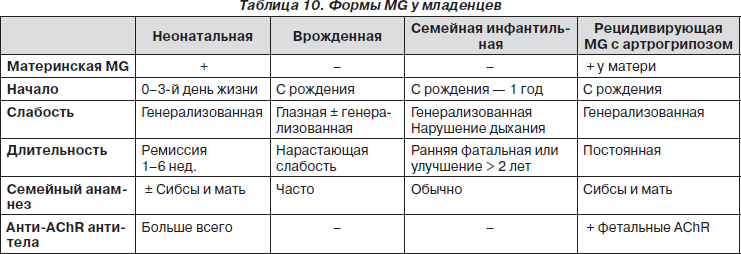
ЭНМГ-признаки: патологическое затухание (декремент) или усиление (инкремент) в зависимости от частоты стимуляции и характера заболевания.
При миастении ритмическая стимуляция ведет к падению амплитуды и площади М-ответа от стимула к стимулу. При тетанизации снижается амплитуда мышечного ответа параллельно со снижением его площади. Декремент отсутствует у младенцев при врожденной МG с эпизодическими апноэ. Декремент не корректируется прозерином при постсинаптическом дефиците ацетилхолинэстеразы (AChE).
Врожденный миастенический синдром (ВМС)
Диагностика ВМС возможна на основе миастенических симптомов, проявляющихся с самого рождения: типичной картины генерализованной гипотонии, слабости глазной, дыхательной, бульбарной и лицевой мускулатуры. У многих новорожденных наблюдается высокое арковидное небо. Снижена реакция зрачков на свет при постсинаптическом дефиците AChE. Редуцированы сухожильные рефлексы у больных с постсинаптическим дефицитом AChE и медленноканальцевыми синдромами; арефлексия может быть при синдроме Lambert — Eaton. Эпизодические апноэ характерны для MG с эпизодическими апноэ, быстроканальцевых симптомов, мутации рапсина, мутации AChR d-субъединицы. Флюктуация симптомов и слабости особенно выражена при MG с эпизодическими апноэ. Артрогрипоз патогномоничен для рецидивирующей врожденной MG, синдрома Эскобара (синдром множественных птеригиумов). Быстрое прогрессирование симптомов наблюдается у новорожденных с медленноканальцевыми синдромами и дефицитом AChE. В семейной истории есть больные родственники с теми же симптомами.
На ЭМГ угасающий ответ потенциала сложного мышечного действия на повторную низкочастотную стимуляцию (2–3 Гц). Антитела к AChR при врожденных миастенических синдромах отсутствуют. Прозериновая проба часто позитивная; негативная проба регистрируется при постсинаптическом дефиците AChE.
Морфологические нарушения в синапсе: уменьшение размеров постсинаптической пластины наблюдается в случае медленноканальцевых синдромов и при дефиците AChR; отсутствие реактивной AchЕ свидетельствует о синдроме дефицита AChE концевых пластин.
Лечение: АХЭ-препараты неэффективны при дефиците AChE; 3,4-Diaminopyridine более эффективен при быстроканальцевых синдромах; Quinidine большей частью эффективен при медленноканальцевых синдромах
Для диагностики некоторых ВМС достаточно простых гистологических и ЭМГ-исследований, а для точной специфической диагностики других необходимы электрофизиологические, структурно-функциональные и иммунохимические исследования.
Исследование врожденных миастенических синдромов:
— клинические данные;
— анамнез, обследование, ответ на ингибитор AСhЕ;
— ЭМГ: обычная игольчатая ЭМГ, повторная стимуляция, одноволоконная ЭМГ;
— серологические тесты (AСhR-антитела, антитела кальциевого канала, тесты на ботулизм);
— морфологические исследования;
— рутинные гистохимические исследования;
— цитохимическое и иммуноцитохимическое определение локализации AСhЕ, AСhR, агрина, b2-ламинина, атрофина (utrophin) и рапсина (rapsyn) на концевой пластине;
— определение размера, формы и конфигурации AСhЕ-реактивных концевых пластин или участков концевых пластин на препарированных мышечных волокнах;
— количественная электронная микроскопия; электронная цитохимия;
— молекулярно-генетические исследования.
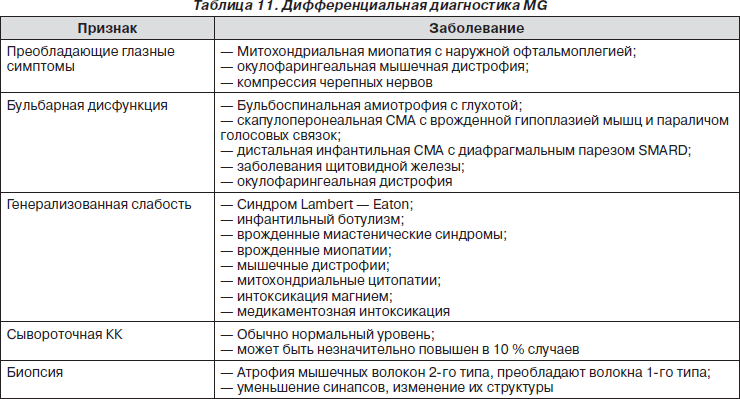
Дифференциальная диагностика МG представлена в табл. 11.
СМГ первично-мышечного уровня
При постепенном развитии двусторонней мышечной слабости в проксимальных отделах конечностей прежде всего необходимо думать о миопатии. Термин «миопатия» в широком смысле понимается как болезнь скелетных мышц. По одной из современных классификаций миопатии подразделяют на врожденные (конгенитальные) структурные миопатии, врожденные мышечные дистрофии, мембранные миопатии с миотоническим синдромом, воспалительные миопатии и метаболические миопатии.
Несмотря на то что ряд форм миотоний сопровождается симптомами прогрессирующей мышечной слабости и гипотрофии, в данной лекции рассмотрение маркеров врожденных миотоний и парамиотоний не предусматривается.
Нарушение моторной интеграции на мышечном уровне характеризуется значительной полифазностью потенциалов, удлинением латенций и декрементом амплитуд М-ответа и Н-рефлекса по данным ЭМГ.
Морфологический дефект, выявляемый в биоптате мышечного волокна, характеризуется атрофией, жировым перерождением и некрозом мышечных волокон с наличием их регенерации, а также разрастанием соединительной ткани эндомизия. При некоторых нозологических формах выявляются специфичные для врожденных доброкачественных структурных миопатий изменения мышечных волокон, такие как центральное расположение ядер или наличие обрамленных вакуолей. Точная диагностика отдельных нозологических форм врожденных миопатий возможна только при проведении молекулярно-генетического анализа, направленного на выявление мутаций в том или ином гене, и в ряде случаев при исследовании концентрации того или иного белка в биоптате мышечного волокна.
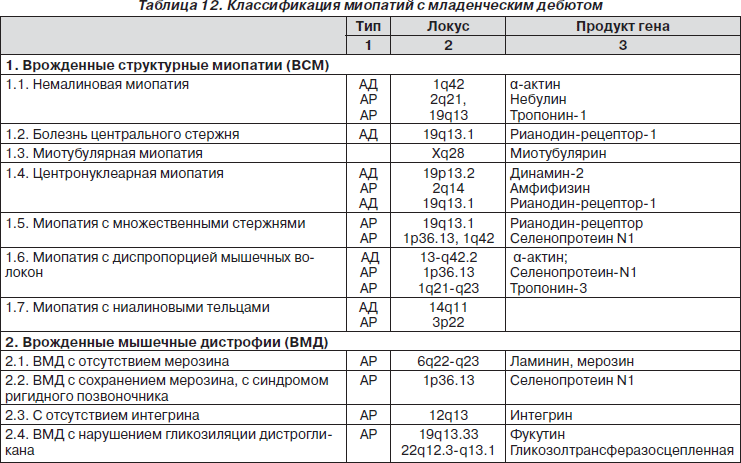
Общепринятой классификации миопатий не существует (табл. 12).
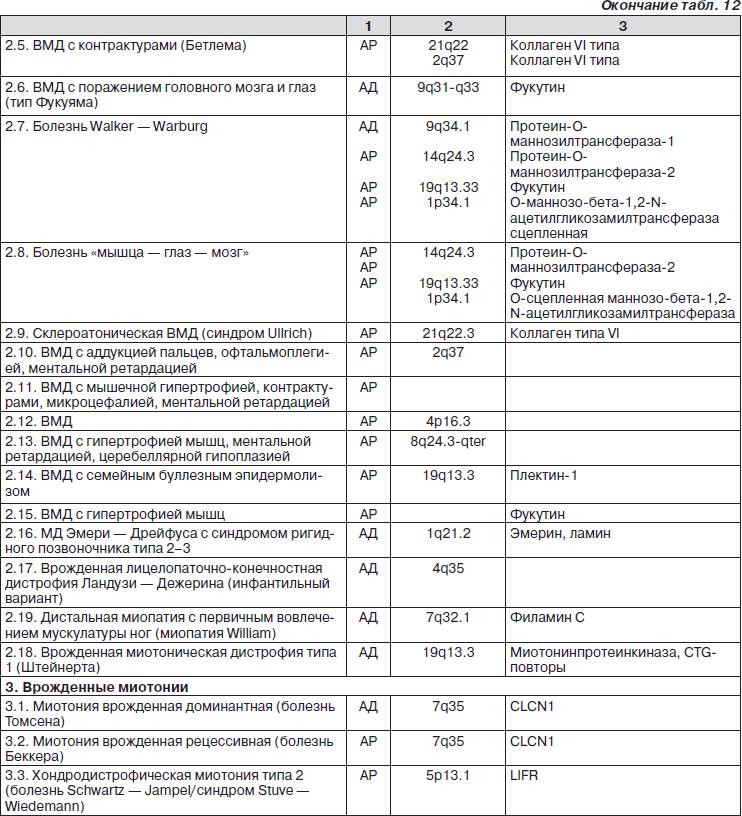
Врожденные структурные миопатии (ВСМ)
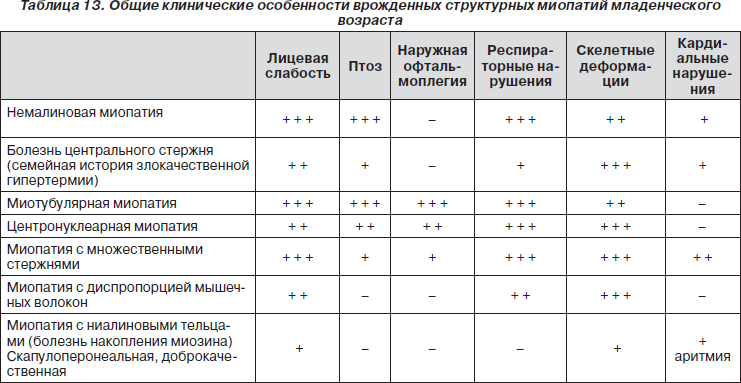
Нарушение функции белков, участвующих в процессах роста, дифференцировке и пролиферации миобластов в раннем эмбриогенезе, приводит к возникновению врожденного структурного дефекта цитоскелета мышц. ВСМ манифестируют в первые месяцы жизни симптомами мышечной слабости, гипотонии, гипорефлексии и задержкой раннего моторного развития при нормальном или умеренно повышенном уровне КФК в крови. Для многих нозологических форм ВСМ характерно вовлечение лицевой и дыхательной мускулатуры, что проявляется ранним респираторным дистрессом, слабым криком и трудностями вскармливания. Часто встречаются краниофасциальные дисморфии, а также деформация позвоночника и контрактуры в крупных суставах (табл. 13). Распознать ВСМ по клинической картине практически невозможно. Для этого используют гистохимические, электронно-микроскопические, биохимические и генетические исследования.
Мышечные дистрофии (МД) — обширная группа моногенных наследственных болезней нервной системы, которые характеризуются нарушением структуры и функции белков сарколеммы мышечного волокна, неспецифическими дистрофическими изменениями в биоптате мышечных волокон и прогрессирующим характером течения заболевания. Каждая из групп МД включает различное число генетически гетерогенных вариантов. Описаны нозологические формы с аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным типами наследования. В рамках одного генетического варианта выделяются аллельные серии, обусловленные различными мутациями в одном и том же гене. Характерными клиническими признаками МД являются симптомы вялого паралича в различных группах мышц без признаков поражения мотонейронов и периферических нервов. Уровень КФК при многих МД значительно повышен.
На ЭМГ выявляется типичный первично-мышечный паттерн, характеризующийся снижением амплитуды М-ответа, усилением интерференции и полифазности потенциала.
В соответствии с существующими представлениями о патогенетических механизмах врожденных МД (ВМД) выделяют две основные группы заболеваний, встречающихся с равной частотой в большинстве европейских популяций: 1) мерозин-негативные, сопровождающиеся первичным или вторичным дефицитом или полным отсутствием локализованного в базальной мембране поперечнополосатых мышц белка мерозина; 2) мерозин-позитивные, при которых концентрация мерозина в мышечных волокнах не отличается от нормы.
Мерозин-негативные формы ВМД подразделяются на несколько типов и включают классический восточный вариант, ВМД Фукуямы, также синдром «мышца — глаз — мозг» и синдром Уолкера — Варбурга, генетическая самостоятельность которых, однако, окончательно не доказана. Клинические симптомы при этих двух формах заболевания имеют много общих черт, характерных для ВМД в целом. Отличительными особенностями мерозин-негативных форм является частое вовлечение в патологический процесс различных структур головного мозга (прежде всего белого вещества полушарий), что клинически проявляется судорогами и умственной отсталостью. Патология глаз включает гипоплазию/атрофию зрительного нерва, колобому, отслойку и нарушение дифференциации сетчатой оболочки, катаракту, дефекты хрусталика, микрокорнеа и микрофтальмию.
При мерозин-позитивных формах ВМД поражение мозга обнаруживается не чаще чем у 10 % больных и, как правило, не сопровождается интеллектуальной недостаточностью. Клиническими признаками мерозин-позитивных форм является наличие деформаций позвоночника и дисморфических черт строения лица. Для ВМД Ульриха характерны кожные изменения (гиперэластичная кожа, папиллярный гиперкератоз, келоидные рубцы, атрофические шрамы, стрии, петехии), синдром ригидного позвоночника, контрактуры, дыхательные расстройства. Фолликулярный (папиллярный) гиперкератоз, келоидные рубцы, атрофические шрамы, кожа как папиросная бумага на локтях и коленях присутствуют также и при ВМД Бетлема. Выраженная слабость лицевой мускулатуры и мышц плечевого пояса в сочетании с потерей слуха, иногда с конвульсиями отмечаются при инфантильной форме МД Ландузи — Дежерина.
Врожденная миотоническая дистрофия проявляется когнитивными нарушениями, респираторным дистрессом, выраженной генерализованной слабостью, арефлексией, миотоническими феноменами, которые ассоциируются с мультисистемными признаками (катаракта, облысение, артрогрипоз, нарушение сердечного ритма, гиперсомния, гипергидроз, гипогонадизм, гипергликемия). МРИ: тотальная гипоплазия серого и белого вещества в лобных и теменных зонах, аномалия таламуса, гиппокампа, мозолистого тела, что коррелирует с когнитивными расстройствами.
Метаболические миопатии с младенческим дебютом
Метаболические миопатии подразделяются на первичные, которые обусловлены наследственным нарушением обмена, и вторичные, которые возникают вследствие интоксикации, соматических и эндокринных заболеваний, транзиторных метаболических расстройств.
Первичные метаболические миопатии — заболевания, которые возникают как проявления болезней лизосомального накопления с сопутствующими дефектами метаболизма мышечного гликогена или липидов. Встречаются при дефиците карнитина, сопутствуют митохондриальным и другим наследственным болезням с нарушением клеточной биоэнергетики.
Эти нарушения приводят к развитию трех основных клинических синдромов: 1) прогрессирующей мышечной слабости с гипотонией; 2) острой рецидивирующей мышечной дисфункции с непереносимостью физической нагрузки и острым распадом мышц и/или миоглобинурией, иногда сопровождаемой крампи; 3) сочетанию двух вышеперечисленных синдромов с мультиорганным поражением.
Причины вторичных метаболических миопатий представлены в табл. 14.

Эндокринопатии: гипотиреоз — синдром Kocher — Debre — Semelaigne (характеризуется холодной, бледной, сухой кожей; отсутствием активности, запорами, макроглоссией, хриплостью голоса, брадикардией, отечностью мышц, снижением ахилловых рефлексов. Нередко сопровождается парестезиями, атаксией, крампи, запястным туннельным синдромом); гипертиреоз (характерны проксимальная мышечная слабость, потливость, тахикардия, тремор, горячая кожа, непереносимость жары, диарея, глазные и пирамидные симптомы); гиперпаратиреоз (отличается истинной миопатией с мышечными атрофиями, эмоциональной лабильностью, запорами). При этих состояниях повышен уровень КК.
Патология надпочечников. Эндогенное повышение в крови уровня кортикостероидов может вызывать резкую мышечную слабость и уменьшение мышечной массы. Недостаточность надпочечников часто сопровождается вялостью мышц и их слабостью, несмотря на сохранение мышечной силы при объективном исследовании.
Патология гипофиза. В отдельных случаях акромегалия сопровождается увеличением мышц. Встречается и мышечная слабость миопатического типа, однако слабость эта скорее является результатом сопутствующей эндокринной патологии или невропатии. Мышечная же слабость при панпитуитаризме, вероятно, возникает вследствие одновременно существующей недостаточности надпочечников и щитовидной железы.
Сахарный диабет. Слабость проксимальных мышц у больных сахарным диабетом обычно является результатом невропатии. Обнаружение же при ЭМГ или биопсии мышцы признаков миопатии, а также повышение активности сывороточной КК обычно указывают на какое-то сопутствующее заболевание.
Витаминная недостаточность. Синдром мальабсорбции, особенно в раннем детстве, может привести к миопатии, связанной с недостаточностью витамина Е. При миопатиях другого генеза витамин Е обычно не применяют для лечения больных. Дефицит же витамина D, независимо от того, возникает ли он в связи с его сниженным употреблением, с нарушением его абсорбции или обусловлен патологией почек, может привести к хронической мышечной слабости; болевые ощущения при этом скорее отражают патологию костей. Недостаток других витаминов обычно не вызывает миопатии.
Другие метаболические расстройства. Такие системные заболевания, как злокачественные новообразования, хроническая дыхательная, сердечная, печеночная или почечная недостаточность, часто сопровождаются существенным уменьшением мышечной массы и мышечной слабостью. При этом тесты на мышечную силу остаются близкими к норме. Признаков мышечного заболевания, как правило, нет. Мышечная слабость в таких случаях может быть обусловлена нарушениями в электролитном обмене, в частности хроническими гипокалиемией, гиперкальциемией или гипокальциемией различной этиологии.
Лекарственная миопатия возникает и при длительном лечении кортикостероидами. В этом случае весьма характерна слабость проксимальной мускулатуры. Прием указанных препаратов несколько раз в день вызывает большую мышечную слабость, чем однократный их прием утром. Одноразовое в течение дня их применение или прием этой дозы через день в большей мере щадит мышечную систему. В подобных ситуациях в пользу вызванной кортикостероидами мышечной слабости свидетельствуют нормальная активность сывороточной КК, нормальная (или с минимальными миопатическими отклонениями) ЭМГ и наличие в мышечном биоптате атрофии мышечных волокон II типа.
В некоторых случаях токсическое воздействие может носить катастрофический характер, вызывая рабдомиолиз и миоглобинурию. Весьма опасно назначение вальпроатов в случае митохондриальных заболеваний, дефицита карнитинпальмитоилтрансферазы, некетотической гиперглицинемии, гипераммониемии, что может привести к усилению клинических проявлений энцефаломиопатии, возникновению синдрома Рейе, рабдомиолизу. Очень серьезным осложнением медикаментозной терапии является злокачественная гипертермия, которая возникает у предрасположенных к ней лиц после применения общих анестетиков и деполяризующих мышечных релаксантов. Используемый при местной анестезии лидокаин может также спровоцировать названное осложнение.
Лабораторные тесты, нейрофизиологические и радиологические методы
Рутинная лабораторная диагностика у новорожденного с СМГ направлена на исключение инфекционных заболеваний и септического процесса. Исследуют культуру крови, мочи, ликвора, определяют уровень электролитов и функцию печени (аммиак, глюкоза, кальций, магний и креатинин); оценивают развернутый анализ крови и мочи. Если присутствует гепатоспленомегалия и кальцинаты на НСГ, необходимо верифицировать TORCH. Если предполагается центральный уровень СМГ, практикующий специалист должен искать генетические и метаболические причины.
Кариотипирование показано, когда присутствует несколько существенных дисморфических признаков. Молекулярное генетическое исследование обеспечивает быструю диагностику. Однако специфическое исследование необходимо выбирать соответственно клиническим признакам у младенца.
Если клиническое обследование предполагает мультисистемное вовлечение (СМГ «плюс»), проводят скрининг, выявляющий врожденные ошибки метаболизма (табл. 6, 7).
В случае ацидоза исследуют аминокислоты в крови и органические кислоты в моче (аминоацидопатии и органические ацидемии), лактат (нарушение обмена углеводов, митохондриальные болезни), пируват, аммиак (дефекты цикла мочевины) и ацилкарнитиновый профиль (органические ацидемии, нарушение обмена жирных кислот).
Длинноцепочечные жирные кислоты и плазмалоген специфичны для пероксисомных нарушений. Уровень КК и соотношение концентрации ацилкарнитина/карнитина помогают в диагностике мышечных дистрофий и дефицита карнитина. Список нейрометаболических нарушений, ассоциированных с СМГ, огромен и находится вне компетенции этой лекции. Уровень КК повышен при миодистрофиях, но остается нормальным при СМА и многих структурных миопатиях. Исследование ДНК полезно при миотонической дистрофии и СМА.
Потенциально полезными в диагностике СМГ являются электрофизиологические исследования, которые выявляют патологические признаки поражения нервов, синапсов и мышц. За исключением нескольких миопатий, полученные нормальные данные ЭМГ свидетельствуют о том, что гипотония — центрального происхождения. Биопсия мышц и нервов с иммуногистохимическим анализом и электронной микроскопией является методом выбора для дифференцирования структурных миопатий и мышечных дистрофий. Если биопсия показывает специфические нарушения, это может подтвердить диагноз.
Нейровизуализация — ценный инструмент для обнаружения повреждений головного мозга. Магнитно-резонансное исследование выявляет структурные аномалии, дефекты нейрональной миграции. Анормальные сигналы в базальных ганглиях свидетельствуют в пользу митохондриальных болезней, а дефекты ствола мозга ассоциированы с синдромом Joubert. Глубинные изменения белого вещества могут быть визуализированы при синдроме Lowe, пероксисомных дефектах. Анормальность мозолистого тела может наблюдаться при синдроме Smith — Lemli — Opitz. Гетеротопии визуализируются при врожденной мышечной дистрофии. МР-спектроскопия также может использоваться для выявления болезни обмена веществ.
Основными задачами терапии СМГ являются комплексное наблюдение и коррекция патологических симптомов врачами различного профиля. Медикаментозный подход включает: а) улучшение невральной проводимости (липоевая кислота, галантамин, нейромидин); б) компенсация энергетического дефицита мышечной ткани (АТФ, рибоксин, карнитин, убихинон, цитомак, коэнзим Q10, оротовая кислота, янтарная кислота, цитофлавин, идебенон; фолиевая кислота, витамины В1, В2, В12, РР, В15, Е и особенно их коферментные формы); в) улучшение тканевого метаболизма (метионин, триметабол, неотон, семакс, идебенон, L-аргинин) и периферического кровообращения (актовегин, трентал, никотиновая кислота, мексидол, тиоцетам); г) прокогнитивные препараты (цитиколин, церебролизин, энцефабол, пирацетам, пантокальцин, цереброкурин, кортексин, брейн-комплекс).


