
Журнал «Медицина неотложных состояний» 6 (37) 2011
Вернуться к номеру
Синдром неадекватной секреции антидиуретического гормона (СНАСАГ) (SIADH — syndrome of inappropriate secretion of antidiuretic hormone): новые возможности коррекции
Авторы: СМИРНОВ И.И. Харьковская областная клиническая больница
Рубрики: Семейная медицина/Терапия, Медицина неотложных состояний
Версия для печати
Электролиты — важный компонент внутренней среды живого организма. Ключевой орган, поддерживающий водно-электролитный гомеостаз, — почки. Задержка либо экскреция жидкости и солей осуществляется при помощи сложного механизма, основную роль в регуляции которого играет антидиуретический гормон — вазопрессин. При развитии СНАСАГ регуляция нарушается, что приводит к гипонатриемии. По некоторым оценкам, гипонатриемия — наиболее частое из встречающихся у стационарного больного электролитных расстройств [1], связанное с неблагоприятным прогнозом и удлинением периода пребывания в стационаре [2–4]. Особенно часто пациентов с гипонатриемией можно встретить в палатах интенсивной терапии онкологических, пульмонологических, неврологических и нейрохирургических стационаров. Несмотря на это, гипонатриемия зачастую остается невыявленной и лишенной адекватной коррекции [5]. Возможно, в прошлом недостаток должного внимания к гипонатриемии был связан с отсутствием эффективных лечебных средств. С появлением антагонистов вазопрессина клинические подходы к диагностике и терапии пересматриваются, растет осознание важности целенаправленной коррекции этого метаболического нарушения.
Осмолярность плазмы, одной из главных составляющих которой является натрий, напрямую связана с содержанием в крови вазопрессина. Такая закономерность впервые была продемонстрирована почти 40 лет назад. На приведенном ниже графике (рис. 1) показана зависимость от концентрации вазопрессина как осмолярности, так и снижения объема циркулирующей жидкости, рассчитанного на изотоническое состояние.
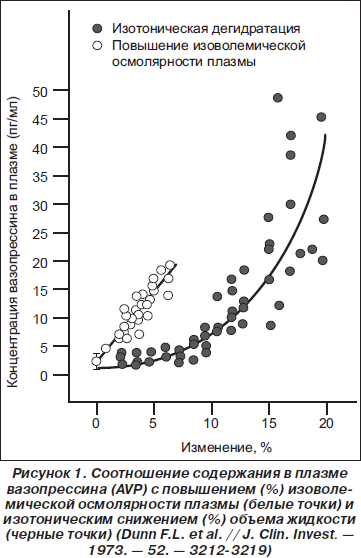
Осморецепторы, реагирующие на перемены осмолярности, расположены непосредственно в клетках супраоптических и паравентрикулярных ядер гипоталамуса, синтезирующих вазопрессин. Однако немаловажную роль в регуляции высвобождения вазопрессина играют также барорецепторы, реагирующие на изменения артериального давления (так называемый неосмотический физиологический механизм регуляции). Чувствительность осморецепторов высока и соответствует изменению осмолярности на 1–2 %. Барорецепторы менее чувствительны, реагируют на 6–10% перемену давления. Несмотря на это, высвобождение вазопрессина в ответ на снижение давления гораздо значительнее, чем при изменении осмолярности. Таким образом, неосмотический физиологический механизм регуляции обладает заметно большим потенциалом и может вызывать повышение уровня вазопрессина в крови, а следовательно — гипонатриемию вследствие резких колебаний артериального давления на фоне любого стрессового состояния [6].
Оба механизма регуляции тесно связаны. Достаточно сказать, что cнижение давления в левом предсердии вследствие гиповолемии и артериальной гипотонии снижает порог возбудимости осморецепторов и увеличивает чувствительность системы осморегуляции секреции вазопрессина. Кроме того, описано большое количество различных патологических состояний, при которых стимулируется секреция вазопрессина. Среди них — злокачественные заболевания мочевого пузыря, предстательной железы, легкого, поджелудочной железы, 12-перстной кишки, лимфомы, лейкозы, мезотелиома, тимома; различные заболевания легких, в том числе пневмония, особенно деструктивные формы, туберкулез, бронхиальная астма; многие заболевания центральной нервной системы (черепно-мозговая травма, острые нарушения мозгового кровообращения, воспалительные и опухолевые заболевания); применение целого ряда лекарственных препаратов. Отдельно следует упомянуть тошноту — чрезвычайно сильный стимулятор секреции вазопрессина. Нередко даже при кратковременной тошноте без рвоты и снижения артериального давления уровень вазопрессина в крови может возрастать на 2–3 порядка [7]. Одно только перечисление факторов, провоцирующих повышение уровня вазопрессина в крови, делает очевидной закономерную частоту развития СНАСАГ.
Точкой приложения действия вазопрессина являются рецепторы типа V2 эпителиальных клеток дистальных канальцев и собирательных трубочек нефрона. Активация этих рецепторов приводит к увеличению числа водных каналов в апикальной мембране, что делает эпителий более проницаемым для воды и обеспечивает ее реабсорбцию. Задержка жидкости ведет к разжижению крови и гипонатриемии. Центральная регуляция жажды обеспечивает ограничение потребления воды, в то время как натрий постоянно выводится с мочой. Важно помнить, что гипонатриемия может иметь место на фоне нормального, пониженного и даже повышенного содержания натрия в организме. Вначале общее содержание натрия может быть сохранным, но при длительном стимулировании высвобождения вазопрессина его запас постепенно истощается и развивается гипонатриемия на фоне нормоволемии и снижения общего количества натрия.
В процессе адаптации центральной нервной системы к отеку на фоне длительной гипонатриемии нейроны для снижения внутриклеточной осмолярности вытесняют в межклеточную среду ионы калия и органические осмотически активные вещества (миоинозитол, аминокислоты — глютамин, таурин), которые впоследствии выводятся с развитием соответствующего внутриклеточного дефицита [8]. Эти метаболические сдвиги являются дополнительными причинами развития клинических проявлений, наблюдаемых при гипонатриемии. В зависимости от быстроты развития и степени выраженности гипонатриемии можно выявить различные клинические проявления, описанные ниже (рис. 2).
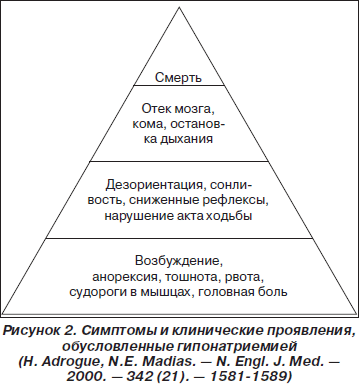
Важно отметить, что чем медленнее и дольше развивается гипонатриемия, тем менее заметны симптомы расстройства. В случаях, когда симптомы выражены в минимальной степени, их набор выглядит неспецифическим, может быть отнесен к проявлениям других болезней и даже к универсальным «возрастным» изменениям. Достаточно сказать, что, по некоторым данным [9], частота случайных падений среди асимптоматических больных с гипонатриемией достигает 21,3 %, а частота переломов — 5,35 %. Высокий показатель переломов, помимо вызванного гипонатриемией нарушения акта ходьбы, связан также с ускорением остеопороза на фоне этого электролитного расстройства.
Для дифференциальной диагностики причин гипонатриемии важным условием является ее сочетание при СНАСАГ с гипернатрийурией на фоне эуволемии. В таком случае требует исключения только гипокортицизм. Если суммарный объем жидкости в организме не изменен, то при гипотиреозе или применении гипотонических растворов гипонатриемия развивается на фоне низкого уровня экскреции натрия с мочой (< 20 ммоль/л). На фоне дегидратации сочетание с гипонатрийурией может иметь место после обильной рвоты, диареи, поражений больших площадей кожи (ожоги), а сопутствующая гипернатрийурия может указывать на предшествовавший прием диуретиков. При наличии гиперволемии причиной гипонатриемии может быть нефротический синдром, а при сниженной экскреции натрия — застойная сердечная недостаточность либо цирроз печени.
Общепринятые сегодня диагностические критерии СНАСАГ предложены советом признанных экспертов (профессора J.G. Verbalis, M. Mannelli, F.C. Luft и доктор E.J. Hoorn). Ключевые диагностические критерии представлены в табл. 1.

Лечение гипонатриемии еще недавно было ограничено в возможностях. Острое понижение содержания натрия в крови можно корректировать при помощи парентерального введения гипертонического раствора натрия хлорида (распространенная мера при лечении аддисонического криза как симптоматическое средство для остановки неукротимой рвоты). Однако при СНАСАГ такого рода лечение дает, как правило, только кратковременный эффект. Для длительного лечения ранее применялось лишь диетическое ограничение потребления жидкости, которое давало слабовыраженный результат, поскольку гипонатриемия вследствие СНАСАГ обычно выявляется на фоне эуволемии.
Заметным событием в эндокринологии стало появление на фармацевтическом рынке первого перорального селективного антагониста V2-рецепторов — толваптана [11, 13]. Блокада V2-рецепторов — непосредственная коррекция нарушения, вызванного чрезмерным количеством антидиуретического гормона. Это воздействие позволяет получить быстрое, устойчивое, клинически значимое повышение содержания натрия в крови, сниженное вследствие СНАСАГ [11, 13, 14]. Новое средство хорошо показало себя в двух первых рандомизированных двойных слепых плацебо-контролированных исследованиях — SALT-1 и SALT-2 (Study of Ascending Levels of Tolvaptan in Hyponatremia) [9]. К сожалению, пока толваптан не зарегистрирован в Украине, но это — дело времени.
На рис. 3 представлен алгоритм лечения гипонатриемии вследствие СНАСАГ, где можно определить место нового лечебного средства. Во всех трех случаях при недостаточном или кратковременном результате терапии возможно назначение антагониста V2-рецепторов в качестве долговременного лечения второго этапа.
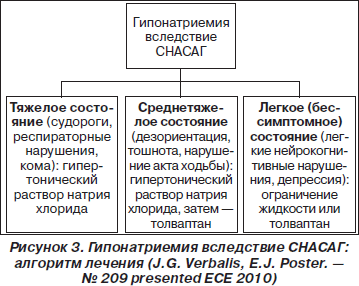
Таким образом, гипонатриемия — нередкое и важное метаболическое расстройство. Хотя не у всех больных с гипонатриемией можно диагностировать СНАСАГ, нарушение секреции вазопрессина выявляется при широком спектре клинических состояний, что позволяет считать СНАСАГ достаточно распространенным обменным нарушением. До недавнего времени арсенал средств при лечении СНАСАГ был в значительной степени ограничен. Появление селективного антагониста V2-рецепторов — толваптана позволяет корректировать гипонатриемию, воздействуя на ключевое звено патогенеза СНАСАГ. Есть надежда, что в скором будущем антагонисты V2-рецепторов позволят решить клиническую проблему, все еще пока обделенную вниманием клиницистов и ограниченную в терапевтических возможностях.
1. Upadhyay A. et al. // Semin. Nephrol. — 2009. — 29 (3). — 227-238.
2. Bissram M. et al. // Intern. Med. J. — 2007. — 37. — 149-155.
3. Sherlock M. et al. // Postgrad. Med. J. — 2009. — 85. — 171-175.
4. Gill G. et al. // Clin. Endo. — 2006. — 65. — 246-249.
5. Hoorn E.J. et al. // Nephrol. Dial. Transplant. — 2006. — 21. — 70-76.
6. Goldberg M. et al. // Med. Clin. North. Am. — 1981. — 65 (2). — 251-269.
7. Robertson G.J. // Harrison’s Endocrinology. — 2006. — 65-69.
8. Schrier R.W., Berl T. // Manual of Nephrology. — 2009. — 27-28.
9. Schrier R.W. et al. // N. Engl. J. Med. — 2006. — 355 (20). — 2099-2112.
10. Ellison D.H. et al. // N. Engl. J. Med. — 2007. — 356. — 2064-2072.
11. Verbalis J.G. et al. // Am. J. Med. — 2007. — 120 (11A). — S1-S21.
12. Janicic N., Verbalis J. // Endocrinol. Metab. Clin. N. Am. — 2003. — 32. — 459-481.
13. Yamamura Y. et al. // J. Pharmacol. Exp. Ther. — 1998. — 287 (3). — 860-867.
14. Hirano T. et al. // J. Pharmacol. Exp. Ther. — 2000. — 292 (1). — 288-294.