
Журнал «Медицина неотложных состояний» 7-8 (38-39) 2011
Вернуться к номеру
Синдром полиорганной недостаточности: «шоковое легкое»
Авторы: Никонов В.В., Павленко А.Ю., Белецкий А.В. Харьковская медицинская академия последипломного образования
Рубрики: Семейная медицина/Терапия, Медицина неотложных состояний
Версия для печати
В предыдущей лекции упоминалось, что при синдроме полиорганной недостаточности (СПОН) поражение систем органов происходит одновременно, однако клинические проявления имеют определенную последовательность. Согласно данным отечественных и зарубежных авторов, при критических состояниях система внешнего дыхания подвергается разрушительному воздействию «цитокиновой бури» в первую очередь.
Начиная с 60-х годов прошлого века разными авторами был описан синдром прогрессирующей острой дыхательной недостаточности, возникающий у больных с острой кровопотерей, тяжелой механической травмой, сепсисом и т.д. Патоморфологическая картина в легких при аутопсии характеризовалась неравномерным кровенаполнением микроциркуляторного русла (вследствие стаза и микротромбоза), экстравазацией плазмы в интерстиций и просвет альвеол, десквамацией респираторного эпителия, деструкцией альвеолоцитов II порядка с формированием гиалиноподобных (фибриновых) мембран и очагов ателектаза. Таким образом, клинические наблюдения и вышеописанные патоморфологические изменения в легких позволили Т. Burford и В. Burbank в 1944 году выделить отдельный клинико-анатомический синдром, назвав его синдромом «влажных легких» [1]. В 1963 году М. Nickerson et al. было обнаружено, что данный синдром наиболее часто встречается при различных шоковых состояниях, и этот патологический процесс был переименован в синдром «шоковых легких» [2]. Впоследствии за ним укрепилось название «респираторный дистресс-синдром взрослых» (adults respiratory distress syndrome — ARDS), предложенное D.G. Ashbaugh et al. (1967). Авторы описали 12 больных с клинической картиной острой дыхательной недостаточности, которая проявлялась диффузным цианозом, резистентным к оксигенотерапии, сниженной растяжимостью (комплайенсом) легких и появлением двусторонних диффузных инфильтратов на рентгенограммах легких [3]. В 1994 году на Американо-Европейской согласительной конференции данный синдром был определен как острый респираторный дистресс-синдром (ОРДС) (аcute respiratory distress syndrome — ARDS) и установлены современные критерии его диагностики [4]. Однако по мере изучения причин развития и патогенеза ОРДС возникла концепция, согласно которой под ним стали понимать крайнее проявление более широкого процесса, именуемого «острым повреждением легких» (ОПЛ) (acute lung injury — ALJ). Было предложено выделять две формы данного заболевания:
1) острое повреждение легких, которое включает в себя как начальный, более легкий, этап заболевания, так и наиболее тяжелые формы;
2) острый респираторный дистресс-синдром, являющийся наиболее тяжелой формой заболевания, т.е. крайним проявлением ОПЛ.
Таким образом, любой ОРДС можно отнести к ОПЛ, но не все формы ОПЛ являются ОРДС. ОПЛ определяется как воспалительный синдром, связанный с повышением проницаемости альвеолокапиллярной мембраны и ассоциированный с комплексом клинических, рентгенологических и физиологических нарушений, которые не могут быть объяснены наличием левопредсердной или легочной капиллярной гипертензией (но могут с ней сосуществовать). Фактически ОПЛ — результат синдрома системного воспалительного ответа, который сопровождается нарушением целостности альвеолокапиллярной мембраны. Клиническим проявлением этих нарушений является развитие некардиогенного отека легких. В табл. 1 представлены рекомендованные критерии ОПЛ и ОРДС.

Таким образом, ОПЛ характеризуется прогрессирующей гипоксемией вследствие внутрилегочного шунтирования крови, двусторонней инфильтрацией легочных полей на фронтальной рентгенограмме грудной клетки, быстрым снижением податливости легочной ткани, легочной гипертензией при отсутствии признаков левожелудочковой недостаточности.
В 1988 году J.F. Murray et al. предложили шкалу тяжести повреждения легких (Lung Injury Score — LIS), которая широко используется в настоящее время в отношении больных, находящихся как на самостоятельном дыхании, так и в условиях проводимой респираторной терапии (табл. 2) [7].
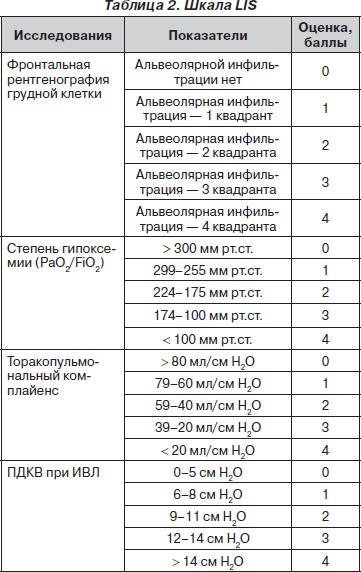
Общая сумма баллов делится на число исследованных компонентов и производится оценка: 0 — повреждения легких нет, 0,25–2,5 — умеренное повреждение легких (летальность составляет 40–41 %), > 2,5 — тяжелый синдром острого повреждения легких (летальность — 58–59 %).
Острое повреждение легких и острый респираторный дистресс-синдром являются практически обязательными компонентами синдрома полиорганной недостаточности у больных в критических состояниях. ОПЛ возникает на основе диффузного повреждения эндотелия легочных капилляров под воздействием экзогенных и эндогенных факторов. Исходя из вышесказанного, ОПЛ может быть следствием как непосредственной альтерации легочной паренхимы (торакальная травма с контузией легких, аспирация желудочного содержимого, инфекционные заболевания легких, утопление и т.п.), так и опосредованного повреждения легких, связанного с внелегочными заболеваниями (сепсис, гиповолемический шок, массивная гемотрансфузия, острый панкреатит, перитонит и т.д.) [5–7].
Несмотря на достигнутый прогресс в понимании этиопатогенеза и лечении ОПЛ, все же летальность при этом синдроме остается довольно высокой и на сегодняшний день составляет от 40 до 60 %. В наиболее общей форме патогенез ОПЛ представлен на рис. 1.
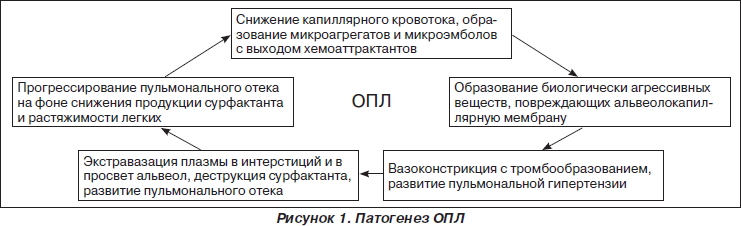
В начальной фазе ОПЛ на фоне замедления кровотока в микроциркуляторном русле легких и образования тромбоцитарных микроагрегатов происходит адгезия и активация нейтрофилов. Последние через поврежденный эндотелий легочных капилляров проникают в просвет альвеол. Нейтрофилы, эндотелиоциты и альвеолярные макрофаги продуцируют цитокины, протеазы (эластаза, коллагеназа) и другие вещества, которые повреждают альвеолокапиллярную мембрану. Дальнейшее патологическое воздействие оказывают продукты каскада комплемента, лизосомальные ферменты и биогенные амины, продукты деградации фибриногена и метаболизма арахидоновой кислоты с появлением в циркулирующей крови простагландинов PgE2 и PgF2, лейкотриенов, тромбоксана, фактора активации тромбоцитов. Эйкосаноиды увеличивают проницаемость мембраны, вызывают бронхиоло- и вазоспазм, усиливают тромбообразование. Активация свободнорадикальных реакций дополняет повреждающее действие цитокинов и эйкосаноидов на клеточные мембраны. В конечном счете вследствие нарушения целостности альвеолокапиллярной мембраны развивается некардиогенный отек легких. В острой фазе синдрома легочная паренхима представляет мозаику воздушных, коллабированных и отечных участков. Перфузия невентилируемых областей является причиной легочного шунта, который может составлять более 60 % (норма 3–7 %) сердечного выброса и являться главной причиной артериальной гипоксемии. В результате вышеописанных патологических изменений возникает несоответствие между вентиляцией и перфузией, которое приводит к возникновению легочного шунта и тяжелой артериальной гипоксемии. Нарушение продукции сурфактанта на фоне альвеолярного отека, воспаления и фиброза приводит к снижению комплайенса легких и повышению «энергетической цены» дыхания.
Исследование механогенеза и лечебно-диагностических аспектов ОПЛ, проведенное на базе кафедры неотложных состояний и медицины катастроф Харьковской медицинской академии последипломного образования, послужило стимулом к разработке концептуальной схемы каскада легочного повреждения при торакальной травме, осложненной легочной контузией (рис. 2). Исходя из этой схемы, основными причинами дыхательных расстройств у пострадавших с закрытой травмой грудной клетки (ЗТГК) следует считать: болевой фактор, первичное повреждение легких вследствие контузии, интраплевральные травматические объемы и повреждение костного каркаса грудной клетки с флотацией участка грудной стенки на месте поражения.
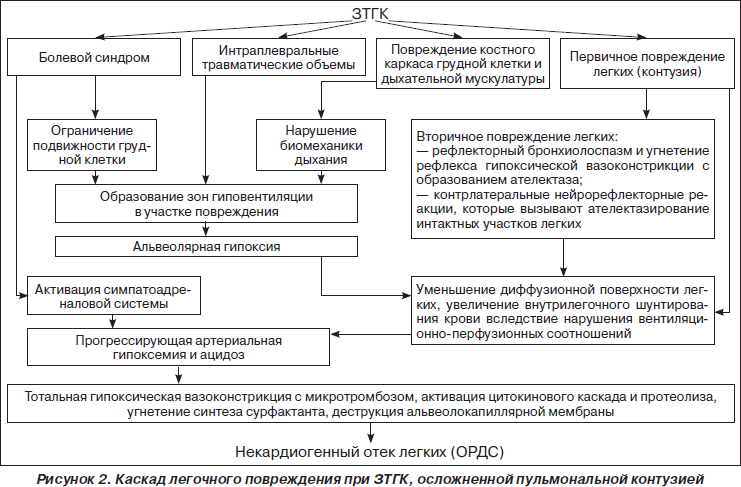
У пострадавших с ЗТГК без ушиба легких вследствие выраженного болевого синдрома возникает ограничение подвижности грудной клетки, нарушение дренажной функции бронхов и склонность к региональному бронхиолоспазму, который способствует образованию зон гиповентиляции на стороне повреждения. Альвеолярная гипоксия, обусловленная гиповентиляцией отдельных участков легких, приводит к рефлекторной вазоконстрикции и перераспределению кровотока в пользу регионов с достаточной вентиляцией. Эта компенсаторная реакция устраняет вентиляционно-перфузионный диссонанс и способствует улучшению газообмена в легких. Однако если региональная гиповентиляция сохраняется продолжительное время, данный компенсаторный механизм становится патогенетическим. Микротромбоз на фоне вазоспазма приводит к деструкции альвеолокапиллярной мембраны, уменьшению диффузионной поверхности и усилению гидратации легочной паренхимы.
У пострадавших с ЗТГК, осложненной ушибом легких, нарушения газообмена более выражены вследствие первичного поражения легочной паренхимы, уменьшения площади диффузионной поверхности, снижения вентиляционно-перфузионных соотношений и прогрессирования интрапульмонального шунта.
Независимо от основного заболевания ургентная постановка диагноза ОПЛ/ОРДС должна осуществляться на основании следующих критериев:
— наличие триггерных факторов: сепсис, политравма, шок, перитонит, пневмония и т.п.;
— балльная оценка по шкале LIS:
-для ОПЛ — от 0,25 до 2,5;
-для ОРДС — более 2,5;
— наличие от двух и более клинических проявлений синдрома системного воспалительного ответа: температура тела больше 38 оС или менее 36 оС; частота сердечных сокращений (ЧСС) более 100 в минуту; частота дыханий более 20 в минуту или PaO2 < 32 мм рт.ст.; лейкоцитоз более 12 000 в мм3, или лейкопения менее 4000 мм3, или наличие более 10 % незрелых форм нейтрофилов.
Наблюдение за динамикой изменений механических свойств легких, рентгенологической картины органов грудной клетки и респираторных показателей позволяет предложить клиническую классификацию стадий ОПЛ/ОРДС, являющуюся производной от шкалы J.F. Murray (1988) [8]:
I стадия — повреждения;
II стадия — субкомпенсированная (мнимого благополучия);
III стадия — прогрессирующая дыхательная недостаточность;
IVстадия — терминальная.
На основании данной классификации может быть получена целостная картина стадийной диагностики ОПЛ/ОРДС.
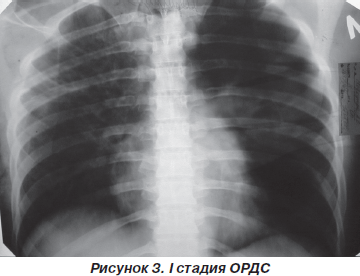
I стадия (24–48 часов от момента действия повреждающего фактора). Состояние больных средней степени тяжести. Клиническая картина и жалобы соответствуют основному заболеванию. Гемодинамика стабильная. При оценке органов дыхания возможно умеренное тахипноэ до 22–26 дых/мин. Аускультативно выслушивается жесткое дыхание, иногда в сочетании с единичными сухими хрипами.
При рентгенологическом исследовании изменения в легких обычно не выявляются. В 20–30 % случаев определяется усиление легочного рисунка (рис. 3).
Газы крови — артериальная гипоксемия, устраняемая ингаляцией кислорода (PaO2/FiO2 < 300 мм рт.ст.), легкая гипокапния (PaCO2 = 33–36 мм рт.ст.). В ряде случаев данная стадия не имеет дальнейшего развития и дыхание восстанавливается без выраженных повреждений легких.
II стадия (48–72 часа от момента действия повреждающего фактора). Состояние больных от средней тяжести до тяжелого. В психологическом статусе часто отмечается эйфория, сменяющаяся беспокойством и негативизмом. Гемодинамика стабильная, возможна синусовая тахикардия.
Обращает на себя внимание выраженная одышка с участием вспомогательной мускулатуры в акте дыхания на фоне стабильного состояния больного. В легких при аускультации выслушивается жесткое дыхание в сочетании с сухими хрипами, а в 25–30 % случаев — зоны ослабленного дыхания, иногда в нижнезадних отделах — влажные хрипы.
На фронтальной рентгенографии органов грудной клетки наблюдаются мелкоочаговые тени по всем легочным полям (рис. 4).
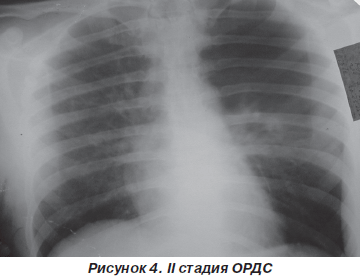
Газы крови — артериальная гипоксемия (PaO2 = = 60–70 мм рт.ст.), резистентная к ингаляции кислорода (PaO2/FiO2 < 200 мм рт.ст.) и выраженная гипокапния (PaCO2 = 30 мм рт.ст.). Увеличение легочного шунта в этой фазе достигает 10–15 % от минутного объема сердца.
III стадия. Состояние больных очень тяжелое. Психомоторное возбуждение сменяется угнетением сознания от оглушения до сопора. Отмечается выраженная тахикардия, артериальное давление (АД) остается нормальным или повышенным, центральное венозное давление (ЦВД) постепенно увеличивается. Наиболее характерный клинический и патофизиологический феномен — зависимость больного от кислорода. Независимо от основного заболевания у всех пациентов наблюдается клиника тяжелой острой дыхательной недостаточности: диффузный цианоз, устраняемый на фоне искусственной вентиляции легких (ИВЛ) с FiO2 = 60–90 %. В легких при аускультации выслушиваются разнокалиберные сухие и влажные хрипы; в 25–30 % случаев — зоны амфорического дыхания. Из трахеи санируется скудная слизистая мокрота.
При рентгенологическом исследовании определяются множественные средне- и крупноочаговые тени с тенденцией к слиянию на фоне снижения интенсивности легочного рисунка («снежная буря»), а в 10–15 % случаев выявляется выпот в плевральных полостях (рис. 5).
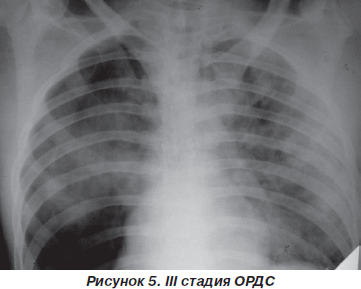
Газовый состав крови — выраженная артериальная гипоксемия (PaO2 = 50–60 мм рт.ст.), резистентная к ИВЛ и оксигенотерапии (PaO2/FiO2 < 175 мм рт.ст.), гипокапния сменяется умеренным повышением РаСО2 до 45 мм рт.ст., метаболический ацидоз. Легочный шунт достигает 20–30 % минутного объема сердца.
IV стадия. Состояние больных крайней степени тяжести или терминальное. Сознание нарушено от сопора до комы. Артериальная гипотензия, требующая применения инотропной поддержки и вазопрессоров; стойкая тахикардия, в дальнейшем переходящая в брадикардию и асистолию; ЦВД может повышаться. Нарушение общей и органной гемодинамики проявляется мраморностью кожи, похолоданием конечностей, олигурией, признаками ишемии миокарда на ЭКГ. Клиника декомпенсированной острой дыхательной недостаточности, которая сохраняется после перевода больных на ИВЛ с FiO2 = 95–100 % и жесткими параметрами вентиляции.
При аускультации на фоне проводимой ИВЛ выслушиваются множество сухих и влажных хрипов по всем легочным полям и резкое ослабление дыхания в заднебоковых отделах. Из трахеи санируется обильная слизистая или слизисто-гнойная мокрота.
На фронтальной рентгенографии органов грудной клетки определяются затемнения долей и сегментов легких (50–52 %) и синдром воздушной бронхографии (48–50 % случаев) (рис. 6).
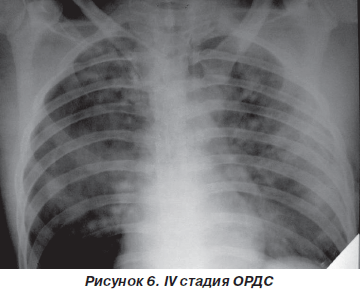
Газовый состав крови — прогрессирование артериальной гипоксемии (РаО2 < 50 мм рт.ст.), резистентной к ИВЛ с ПДКВ (PaO2/FiO2 < 100 мм рт.ст.), нарастание гиперкапнии (РаСО2 > 50 мм рт.ст.). Легочный шунт иногда достигает 50–60 % минутного объема сердца. Развивается метаболический и респираторный ацидоз со снижением рН артериальной крови до 7,10–7,15, усугубляются фатальные расстройства других органов и систем.
Тактика интенсивной терапии ОПЛ зависит от тяжести состояния больного и должна преследовать следующие цели:
1. Этиотропная терапия, направленная на борьбу с заболеванием, вызвавшим развитие ОПЛ.
2. Проведение респираторной терапии с целью поддержания адекватного газообмена (оксигенотерапия, комбинация режимов ИВЛ).
3. Санация трахеобронхиального дерева с применением муко- и бронхолитиков.
4. Оптимизация легочного кровотока и противоотечная терапия (инотропная терапия, нитраты, кортикостероиды, ингибиторы протеаз, салуретики на фоне контролируемой инфузионной терапии).
5. Деэскалационная антибактериальная терапия, коррекция синдрома эндогенной интоксикации.
6. Профилактика геморрагической гастроэнтеропатии (антациды, Н2-блокаторы, М-холинолитики, ингибиторы протонной помпы).
7. Нутритивная поддержка.
Кардинальным вопросом интенсивной терапии ОПЛ является своевременная и адекватная респираторная терапия, которая должна придерживаться концепции безопасной ИВЛ:
— пиковое давление в дыхательных путях не более 35 смН2О;
— дыхательный объем не более 6–8 мл/кг массы тела;
— частота дыхания и минутный объем вентиляции минимально необходимые для поддержания РаСО2 на уровне 30–40 мм рт.ст.;
— скорость пикового инспираторного потока в диапазоне от 30 до 80 л/мин;
— профиль инспираторного потока нисходящий (рампообразный);
— FiO2 — минимально необходимая для поддержания достаточного уровня оксигенации артериальной крови;
— выбор РЕЕР в соответствии с концепцией оптимального РЕЕР, при котором транспорт кислорода к тканям максимальный;
— выбор ауто-РЕЕР — избегать появления высокого ауто-РЕЕР — не более 50 % от величины общего РЕЕР;
— продолжительность инспираторной паузы не более 30 % от продолжительности времени вдоха;
— отношение вдох/выдох — не инвертировать отношение вдох/выдох более 1,5 : 1;
— при десинхронизации больного с респиратором — использование анальгоседации и при необходимости непродолжительной миоплегии, а не гипервентиляции [9, 10].
Поддержание газообмена на различных этапах интенсивной терапии при ОРДС осуществляется с помощью различных вариантов ИВЛ. При тяжелых формах ОРДС наиболее оптимальными режимами является вентиляция по давлению, а не по объему. Индивидуальный выбор параметров и режимов искусственной вентиляции легких, в соответствии с концепцией «безопасной ИВЛ», должен обеспечивать достаточную аэрацию легких и оксигенацию крови без существенных нарушений гемодинамики.
При проведении респираторной поддержки при ОПЛ/ОРДС целесообразно применять кинетическую терапию: прон- и латеропозиции (положение больного на животе и на боку), которая позволяет увеличить индекс оксигенации на 30–40 % от исходного. Однако в процессе использования данного метода могут возникать расстройства центральной гемодинамики, повышение внутричерепного давления и обструкция трахеобронхиального дерева. По данным рандомизированных исследований, кинетическая терапия улучшает оксигенацию, но не увеличивает выживаемость пациентов с ОПЛ [11, 12].
В заключение хотелось бы в качестве примера привести два клинических случая течения ОПЛ у больных с сочетанной травмой.
Больная Ж., 22 года, поступила в стационар после падения с высоты четвертого этажа. Диагноз: острая тяжелая черепно-мозговая травма, ушиб головного мозга тяжелой степени с очагами контузии корковых отделов лобных долей и передних отделов мозолистого тела, субарахноидальное кровоизлияние; закрытая травма грудной клетки, ушиб легких и сердца; ушиб почек; закрытые переломы обеих седалищных костей. Содержание этилового спирта в крови 2,15‰. При поступлении состояние больной тяжелое. Уровень сознания — глубокое оглушение (10 баллов по шкале комы Глазго (ШКГ)). Функции внешнего дыхания компенсированы, напряжены: ЧДД — 26–28 дыханий в минуту, SpO2 — 90 %. Гемодинамика компенсирована: АД = 110/70 мм рт.ст., PS = 100 ударов в минуту. УЗИ сердца: отмечаются зоны гипокинезии по задней стенке, EF — 40 %. При аускультации над легочной поверхностью отмечается жесткое дыхание, ослабленное в базальных отделах (больше справа). На рентгенограмме органов грудной клетки определяется затемнение нижней доли правого легкого (зона контузии) на фоне усиления легочного рисунка (рис. 7).
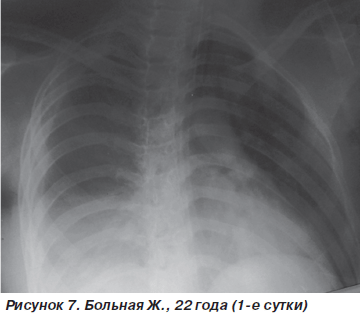
С целью контроля волемии произведена катетеризация подключичной вены по Сельдингеру (ЦВД = = 90 мм вод.ст.), на фоне консервативной терапии начата ингаляция увлажненной воздушно-кислородной смеси (FiO2 = 50–60 %) через маску Вентури. В ходе проведенных лечебных мероприятий состояние больной несколько стабилизировалось (ЧДД — 22–24 дыхания в минуту, SpO2 — 92–94 %, АД = 120/70 мм рт.ст., ЦВД = 50 мм вод.ст., PS = 94–96 ударов в минуту).
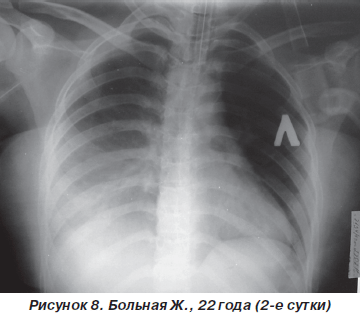
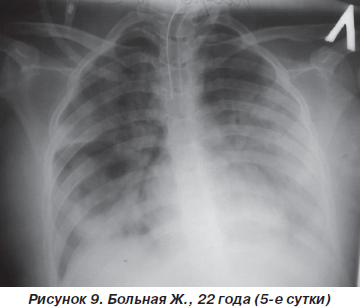
Однако к концу вторых суток состояние больной снова ухудшилось. Уровень сознания — сопор (9 баллов по ШКГ) с периодами психомоторного возбуждения. Тахипноэ до 30–32 дыханий в минуту, SpO2 — 87–89 % на фоне ингаляции увлажненного кислорода (FiO2 = = 90–100 %). Гемодинамические показатели: АД = = 150/90 мм рт.ст., ЦВД = 100 мм вод.ст., PS = 108–112 ударов в минуту. При аускультации легких: жесткое дыхание, ослабленное в средних и нижних отделах (особенно справа), в сочетании с сухими и влажными хрипами. На рентгенограмме органов грудной клетки: затемнение средних и нижних отделов правого легкого, расширение корней легких и усиление сосудистого компонента легочного рисунка (рис. 8). После интубации трахеи больная переведена на контролируемую ИВЛ с анальгоседацией и миоплегией. С целью проведения длительной респираторной терапии и адекватной санации трахеобронхиального дерева на 4-е сутки больной произведена трахеостомия по Бьерку. Однако, несмотря на проводимый комплекс мероприятий интенсивной терапии и комбинаций различных режимов ИВЛ, состояние больной неуклонно ухудшалось. В динамике патоморфологические изменения в легких (рис. 9, 10) коррелировали с клиническим статусом и степенью гипоксемии. Больная скончалась на 12-е сутки на фоне прогрессирующей декомпенсации кардиореспираторных функций.
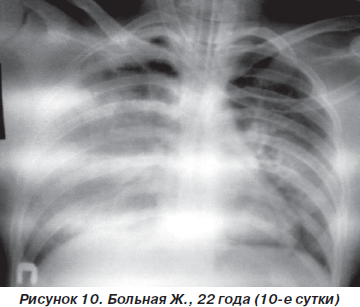
Резюме: данный клинический случай констатирует, что ОПЛ, развившееся вследствие первичной травмы легких, прогностически часто имеет неблагоприятное течение, особенно в сочетании с ушибом сердца и тяжелой черепно-мозговой травмой.
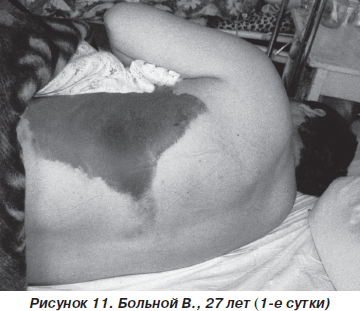
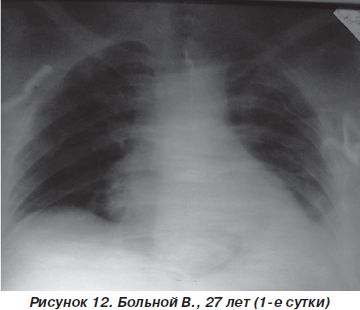
Больной В., 27 лет, пострадал в результате ДТП (водитель), госпитализирован с диагнозом: закрытая травма грудной клетки, переломы III–V и VII ребер слева, ушиб левого легкого, ушиб передней брюшной стенки (рис. 11). При поступлении состояние больного тяжелое. В сознании, жалуется на резкую болезненность в левой половине грудной клетки в покое, нехватку воздуха. ЧДД — 26–28 дыханий в минуту, SpO2 — 90–92 %, АД = 140/80 мм рт.ст., PS = 110 ударов в минуту. При аускультации: над всей поверхностью легких выслушивается жесткое дыхание, резко ослабленное в базальных отделах слева, в сочетании с влажными хрипами. На рентгенограмме органов грудной клетки определяются переломы ребер слева, снижение прозрачности левого легкого вследствие ушиба (рис. 12).
На фоне комплексной интенсивной терапии, включавшей также оксигенотерапию, сеансы гиперинфляции и физиотерапии, больному с целью обезболивания с первых суток проводилась продленная потенцированная субплевральная блокада по методике, разработанной кафедрой (Декларативный патент на полезную модель «Спосіб проведення пролонгованої потенційованої ретроплевральної блокади» (№ 20040705388)).
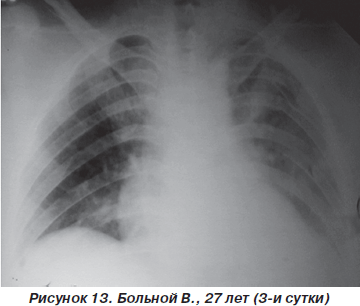
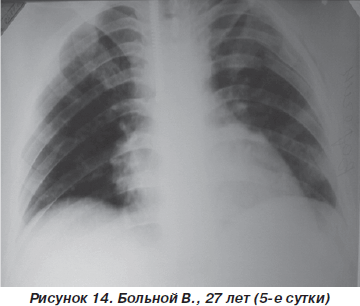
На третьи сутки после травмы у пострадавшего значительно уменшились болевые ощущения, появился продуктивный кашель. ЧДД — 20–22 дыхания в минуту, SpO2 — 92–95 %, АД = 110/10 мм рт.ст., PS = 90–94 удара в минуту. Аускультативно: на фоне жесткого дыхания, ослабленного в средних и нижних отделах слева, единичные сухие хрипы. На рентгенограмме грудной клетки: усиление легочного рисунка, инфильтративные изменения легочной ткани справа в нижних отделах и слева — практически всего легкого (рис. 13). Таким образом, несмотря на некоторое ухудшение рентгенологической картины, дыхательные нарушения у больного оставались компенсированными и не требовали более агрессивных методов респираторной поддержки. Однако на 5-е сутки пребывания в стационаре отмечалась выраженная положительная динамика как параметров оксигенационной функции легких, так и рентгеноморфологических изменений (рис. 14).
Больной выписан из стационара на 20-е сутки в удовлетворительном состоянии.
Резюме: данный клинический случай демонстрирует возможности комплексного подхода в лечении ОПЛ на фоне торакальной травмы, а именно комбинации медикаментозного и физиотерапевтического воздействия, которые позволяют оптимизировать респираторные функции и устранить фатальные расстройства дыхания.
Невзирая на достижения современной медицины, лечение ОПЛ все же остается наиболее актуальной проблемой в контексте полиорганных нарушений. Согласно данным отечественных и зарубежных исследователей, даже такие перспективные методики, как использование искусственных сурфактантов, оксида азота, простациклина, применение жидкостной вентиляции легких и экстракорпоральной мембранной оксигенации крови, не дают однозначно положительных результатов в течении ОПЛ. Вполне вероятно, что в ближайшем будущем мы станем свидетелями новых направлений лечения этого состояния.
1. Burford Т., Burbank B. Traumatic wet lung // J. Thorac. Surg. — 1945. — 14. — P. 415-424.
2. Martin A., Simmonds R., Heisterkamp C. Respiratory insufficiency in combat casualties. Pathologic changesin the lungs of patients dying of wounds // Ann. Surg. — 1969. — 170. — P. 30-38.
3. Ashbaugh D.G., Bigelow D.B., Petty T.L. et al. Acute respiratory distress in adults // Lancet. — 1967. — Vol. 12, № 7511. — P. 319-323.
4. Bernard J.R., Artigas A., Brigham K.L. et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination // Am. J. Res. Crit. Care Med. — 1994. — Vol. 149. — P. 818-824.
5. Глумчер Ф.С. Острый респираторный дистресс-синдром: определение, патогенез, терапия // Мистецтво лікування. — 2004. — № 9(15). — С. 12-17.
6. Флорикян А.К. Хирургия повреждений груди. — Харків: Основа, 1998. — 504 с.
7. Murray J.F., Mattham M.A., Luce J.M., Flick M.R. An expanded definition of the adult respiratory distress syndrome // Am. Rev. Respir. Dis. — 1988. — Vol. 138. — Р. 720-723.
8. Рябов Г.А. Гипоксия критических состояний. — М.: Медицина, 1988. — 287 с.
9. Piantadosi C.A., Schwartz D.A. The Acute Respiratory Distress Syndrome // Ann. Intern. Med. — 2004. — V. 141. — 460-470.
10. Slutsky A.S. The Acute Respiratory Distress Syndrome, Mechanical Ventilation, and the Prone Position // N. Engl. J. Med. — 2001. — V. 345. — 610-612.
11. Gattioni L. et al. Prone-Supine Study group. Effect of prone positioning in acute respiratory distress syndrome // N. Engl. J. Med. — 2001. — V. 345. — 568-573.
12. Pelosy P. et al. Prone position in acute respiratory distress syndrome // Eur. Respir. — 2002. — V. 20. — 1017-1028.