
Газета «Новости медицины и фармации» Антимикробная терапия и пульмонология (408) 2012 (тематический номер)
Вернуться к номеру
Хинолоны, нафтиридоны и фторхинолоны: фармакокинетика и клиническое применение
Авторы: Э.М. Ходош, к.м.н., ассистент кафедры фтизиатрии и пульмонологии ХМАПО
Версия для печати
Моя единственная сила — это мое упорство.
Л. Пастер
Когда появились первые хинолоны, никто не ожидал, что они так стремительно ворвутся в клиническую медицину и будут иметь блестящее будущее. Более 40 лет назад случайно из антималярийного препарата хлорохина была синтезирована налидиксовая кислота. Налидиксовая кислота и созданный в последующем ряд препаратов (розоксацин, оксолиниевая и пиромидиевая кислоты, циноксацин и др.) обладали умеренной активностью в отношении грамотрицательных микробов и плохим всасыванием из желудочно-кишечного тракта (ЖКТ), создавая низкие концентрации в крови, в связи с чем не могли применяться для лечения системных инфекций. Однако благодаря высоким концентрациям, создающимся в моче и фекалиях, они нашли применение для лечения инфекций мочевыводящих путей (МВП) и некоторых кишечных инфекций. Кроме того, в процессе клинического применения выяснилось, что у микробов к ним достаточно быстро развивается резистентность, что также ограничивало их использование в клинике.
Введение пиразинильной цепи в положение 7 ядра нафтиридона или хинолона (например, пипемидиевая кислота) привело к некоторому повышению антимикробной активности в отношении грамотрицательных бактерий, некоторому расширению спектра действия (включая Pseudomonas spp. и некоторые грамположительные микробы).
Принципиально новые перспективы в создании лекарств с повышенной антимикробной активностью появились при введении атома фтора в положение 6. Первым фторсодержащим препаратом был флумехин (1973), создание которого свидетельствовало о возможности повышения активности соединений этого класса в отношении грамположительных микробов.
Реальным достижением в химии фторхинолонов было синтезирование норфлоксацина (фторированного в положении 6 хинолона в сочетании с пиперазинильным кольцом в положении 7): препарат имел активность в отношении широкого спектра грамотрицательных бактерий и некоторых грамположительных микробов. Однако его фармакокинетические свойства недостаточны для применения в качестве средства для лечения системных инфекций. После получения норфлоксацина начались интенсивные исследования по синтезу новых соединений в ряду фторхинолонов: начиная с 1978 года (патентование норфлоксацина) в последующие 3–4 года были получены многие фторхинолоны, часть которых применяется до настоящего времени — пефлоксацин (1979), эноксацин (1980), флероксацин (1981), ципрофлоксацин (1981), офлоксацин (1982).
За последние два десятилетия клинические испытания прошли больше чем 30 препаратов группы хинолонов, нафтиридонов и фторхинолонов. Из них 12 предложены для широкого использования в медицинской практике (табл. 1). Фторхинолоны вместе с цефалоспоринами последних поколений и карбапенемами принадлежат к трем основным классам антимикробных препаратов широкого спектра действия. Одно из главных преимуществ фторхинолонов — наличие таблетированных лекарственных форм с высокой биодоступностью.
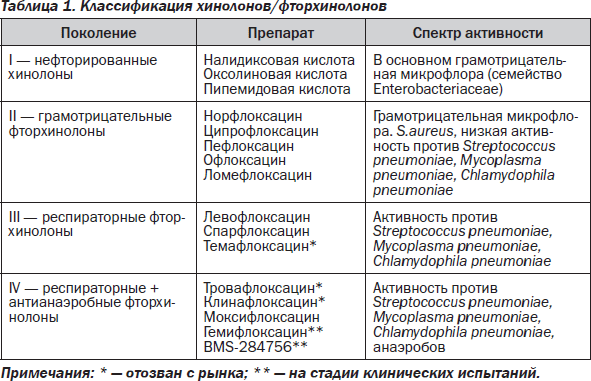
Несмотря на широкий антимикробный спектр и высокую активность, недостатком ранних фторхинолонов является недостаточно высокая активность в отношении грамположительных кокков и некоторых других микробов, являющихся возбудителями респираторных инфекций. Кроме того, отмечается развитие резистентности микробов к фторхинолонам, в частности S.aureus и P.aeruginosa.
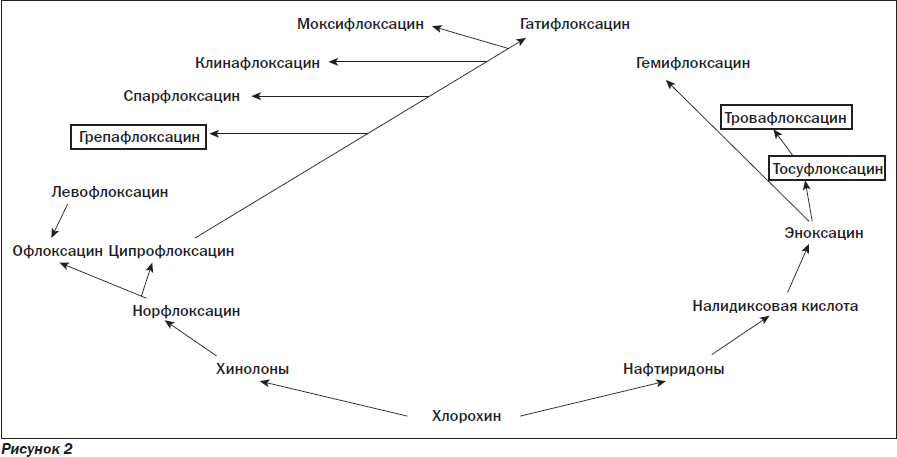
Также в связи с большим количеством побочных эффектов исключены из клинического использования новые фторхинолоны (грепафлоксацин) и новые нафтиридоны (тосуфлоксацин и тровафлоксацин). Новое поколение фторхинолонов расширяется и постоянно пополняется новыми препаратами, к которым относят левофлоксацин и моксифлоксацин. Таким образом, из небольшой группы препаратов, использовавшихся для лечения инфекций мочевыводящих путей, фторхинолоны превратились в один из доминирующих классов антибиотиков.
История развития антибиотиков класса хинолонов, безусловно, зависела от их структуры, которая имеет в своей основе два шестичленных цикла. По химическому строению различают хинолоны (группа СН в восьмом положении) и нафтиридоны (атом азота в восьмом положении). Для фторхинолонов принципиально наличие атома фтора в шестом положении.
Первые хинолоновые препараты — налидиксовая и пиромидиевая кислоты были синтезированы в период 1962–1969 гг. что совпало по времени с созданием ампициллина и гентамицина (табл. 2).
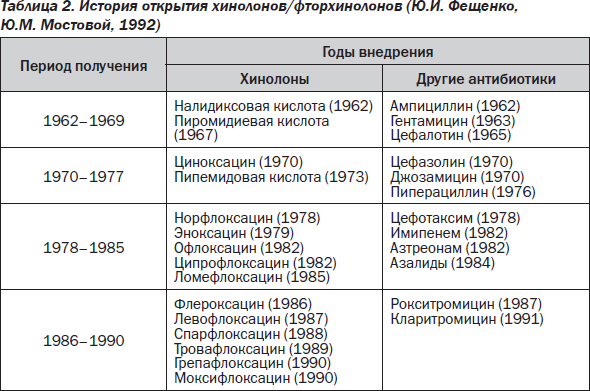
Если ампициллин и гентамицин по праву называют народными антибиотиками, то о первых хинолонах можно упомянуть лишь в чисто историческом аспекте.
Такая же участь постигла хинолоновые препараты, которые были синтезированы в 70-е годы прошлого века. Немногие клиницисты могут вспомнить сегодня такие препараты, как циноксацин или пипемидовую кислоту, зато цефазолин, выведенный на рынок в этот же период, до настоящего времени не утратил своей значимости.
В дальнейшем ситуация изменилась коренным образом. Фторхинолоновые препараты, созданные в конце 70-х — середине 80-х годов, — норфлоксацин, офлоксацин, ципрофлоксацин — заняли достойные места.
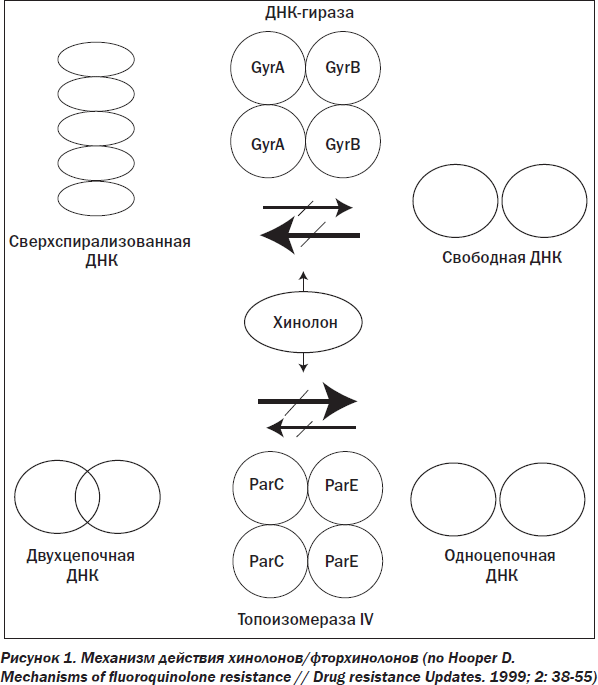
Все хинолоны синтезируются химическим путем, то есть они не являются продуктами жизнедеятельности микроорганизмов. Налидиксовая кислота (4-хинолон-3-карбоновой кислоты) — первый антибактериальный хинолон, который внедрен в клиническую практику в 1963 году. Она подавляет рост грамотрицательных бактерий и в меньшей степени грамположительных микроорганизмов. Налидиксовая кислота — это нафтиридоновое производное, тогда как новые хинолоны, примерно в тысячу раз более активные, представляют собой истинные хинолоновые соединения. Все они связываются с ферментом ДНК-гиразой, участвующим в образовании новых молекул ДНК (рис. 1) (фермент, ответственный за топологическую организацию бактериальной клетки. ДНК-гираза принимает участие в процессах репликации, генетической рекомбинации и репарации ДНК. При блокаде ДНК-гиразы разрушается генетический код бактерий, что приводит к их гибели. Причем они разрушаются в такой степени, что в дальнейшем не способны восстановиться среди антибактериальных средств, как и синтезированные в это время цефотаксим, имипенем, азтреонам).
Исторически первые хинолоны (налидиксовая кислота и близкие к ней по химической структуре и свойствам оксолиновая кислота, а также циноксацин) не создавали системных антибактериальных концентраций и, таким образом, использовались только в качестве мочевых антисептиков на протяжении 20 лет.
Широко распространенным препаратом этой группы является норфлоксацин. В отличие от других фторхинолонов он создает высокие концентрации только в ЖКТ и мочеполовых путях. Биодоступность — 70 %, период полувыведения (t1/2 — 3–4 часа. Интересны показания к применению этого препарата: инфекция МВП, простатит, кишечные инфекции (шигеллез, сальмонеллез), гонорея. «Все жанры хороши, кроме скучного», то есть препарат эффективен против многих грам- отрицательных бактерий, но не частых обитателей респираторного тракта.
Так, налидиксовая кислота угнетает многие грамотрицательные бактерии in vitro в концентрациях 5–20 мкг/мл. Для проявления своего действия in vivo необходимы значительно более высокие концентрации. Большинство штаммов Е.соli чувствительны к этому препарату, как и некоторые штаммы Enterobacter, Klebsiella и Proteus mirabilis. Pseudomonas и большинство положительных микроорганизмов — золотистый стафилококк, пневмококк и энтеробактерии — обычно устойчивы.
Между налидиксовой кислотой, оксолиновой кислотой и циноксацином существует перекрестная устойчивость. Их назначают для приема внутрь, и они почти полностью абсорбируются из желудочно-кишечного тракта, расщепляются в печени на биологически активные и неактивные соединения, которые выводятся почками. Приблизительно 20 % препарата, попавшего в кровь, экскретируется с мочой в активной форме, а 80 % — в виде неактивных конъюгатов с глюкуроновой кислотой. Концентрация активной формы препарата в моче достигает 50–200 мкг/мл.
Наибольшие трудности при применении этих препаратов заключаются в быстром развитии лекарственной устойчивости к ним. Появление устойчивых микроорганизмов при лечении этими препаратами обусловлено как селекцией резистентных мутантов в микробной популяции, так и суперинфекцией лекарственно-устойчивыми организмами других штаммов или видов. Именно в связи с этим обстоятельством их не считают высокоэффективными при инфекциях мочевых путей.
Передача плазмидами резистентности к налидиксовой кислоте не установлена. Перекрестной устойчивости возбудителей к налидиксовой кислоте и другим антимикробным препаратам этой группы нет.
Итак, единственным показанием к применению этих препаратов является инфекция мочевыводящих путей, вызванная колиформными организмами. Доза налидиксовой кислоты (невиграмон, неграм и др.) для взрослых составляет 1 г перорально 4 раза в день в течение 1–2 недель (для детей — 30–60 мг/кг в сутки). Оксолиновая кислота (диоксацин, грамурин и др.) назначается в дозе 0,75 г два раза в день, а циноксацин — в дозе 1 г/день.
Выводящаяся с мочой налидиксовая кислота может вызывать ложноположительную реакцию на глюкозу, но может способствовать и возникновению истинной гипергликемии и глюкозурии. Иногда отмечаются диспептические явления, кожные высыпания, фотосенсибилизация, нарушения зрения, различные явления со стороны центральной нервной системы (ЦНС), включая судороги. Хинолоны ингибируют окислительные ферменты печени и могут усиливать действие лекарств, метаболизируемых системой цитохрома Р450. В частности, под влиянием хинолонов усиливаются побочные действия, вызываемые теофиллином. Токсическое действие на ЦНС более характерно для оксолиновой кислоты.
Однако новые или, как их еще называют, респираторные фторхинолоны не метаболизируются ферментами системы цитохрома Р450, а значит, не взаимодействуют с варфарином и теофиллином (и в целом характеризуются минимальной степенью лекарственных взаимодействий).
Другой характерной токсилогической особенностью хинолонов является обнаруженное в условиях эксперимента явление артропатии у молодых (растущих) животных, выражающееся в возникновении везикул и эрозий на хрящевой поверхности суставов. До сих пор нет данных о появлении этой патологии у людей, тем не менее хинолоны противопоказаны детям и подросткам (в период формирования скелета), а также беременным и кормящим матерям.
Огромным шагом вперед явился переход от норфлоксацина к группе ципрофлоксацина/офлоксацина. В процессе изучения этой группы препаратов был синтезирован целый ряд производных 4-хинолона. Соединения этой группы оказались активными антибактериальными средствами, причем особенно активны соединения, содержащие в положении 7 хинолонового ядра незамещенный или замещенный пиперазиновый цикл, а в положении 6 — атом фтора. Эти соединения названы фторхинолонами (хинолоны второго поколения).
Раличают монофторированные соединения (ципрофлокацин, офлоксацин, эноксацин, пефлоксацин, амифлоксацин, руфлоксацин), ди- (дифлоксацин, амефлоксацин) и трифторированные соединения (флероксацин, темафлоксацин).
Итак, вторая волна развития хинолонов связана с появлением фторированных соединений с низкой токсичностью и гораздо более высокой активностью в отношении широкого спектра грамотрицательных микроорганизмов и некоторых грамположительных возбудителей (Staphylococcus aureus). Данные препараты практически не действуют на стрептококки. Антихламидийную активность проявляет только офлоксацин. Фторхинолоны обладают улучшенной фармакокинетикой, имеют формы для парентерального введения и, вследствие этого, расширенные показания для применения.
Фармакокинетическая и клиническая значимость респираторных фторхинолонов
Большинство фторхинолоновых препаратов, синтезированных в конце 80-х годов прошлого столетия, стали рассматриваться как конкуренты цефалоспоринов III–IV поколения. Это стало возможным благодаря ряду следующих обстоятельств:
— уникальный среди антимикробных препаратов механизм действия — ингибирование фермента бактериальной клетки — ДНК-гиразы и, по последним данным, топоизомеразы IV;
— чрезвычайно высокая степень бактерицидной активности в отношении большинства чувствительных к ним микроорганизмов. Если бактериостатики типа макролидов и тетрациклинов пригодны преимущественно для лечения легких и среднетяжелых инфекций, то фторхинолоны — для лечения инфекционных заболеваний любой степени, включая тяжелые их формы;
— широкий спектр антимикробного действия, в первую очередь в отношении грамотрицательных аэробных бактерий, ряда анаэробов, атипичных микроорганизмов (хламидии, микоплазмы, легионеллы), микобактерии;
— высокая биодоступность при приеме внутрь, хорошее проникновение в ткани, клетки макроорганизма, способность создавать концентрации, близкие и превышающие сывороточные;
— уничтожение возбудителей инфекционных заболеваний минимальным высвобождением различных компонентов бактериальной клетки (эндотоксинов), что редко приводит к развитию эндотоксического шока;
— пролонгированный период полувыведения, наличие постантибиотического эффекта, что позволяет применять ряд препаратов один или два раза в сутки;
— хорошее потенцирующее сочетание с другими группами антибактериальных препаратов (бета-лактамами, аминогликозидами, макролидами, линкосамидами, имидазолами);
— возможность применения в качестве эмпирической терапии при тяжелых инфекциях в стационаре;
— низкая частота резистентности к ним бактерий, хорошая переносимость, небольшая частота побочных эффектов.
Золотым стандартом хинолонов II поколения стал ципрофлоксацин, разработанный в лабораториях компании Bayer AG, зарегистрированный и разрешенный к применению во многих странах мира с 1987 года. С тех пор препарат с большим успехом используется для лечения многих инфекций. Он хорошо всасывается в ЖКТ, его биодоступность составляет 80 %, t1/2 — 4–6 часов. Ципрофлоксацин (ципробай, цифран) обладает рядом уникальных свойств:
— широким спектром действия против клинически значимых аэробных микроорганизмов;
— очень хорошим проникновением в ткани (концентрация препарата в тканях соответствует или превышает концентрацию в плазме);
— быстрым бактерицидным действием на всех стадиях роста бактерий и способностью воздействовать на внутриклеточные микроорганизмы.
Параллельно резистентность к другим антибиотикам обычно не развивается, и ципрофлоксацин успешно применяют для лечения инфекций, вызванных полирезистентными микроорганизмами.
К недостаткам препаратов этого поколения следует отнести низкую активность в отношении пневмококков, хламидий, микоплазм и анаэробов. Самым активным среди хинолонов II поколения против пневмококков и хламидий оказался офлоксацин. Однако он хуже, чем ципрофлоксацин, действует на P.aeruginosa. Препарат практически полностью всасывается в ЖКТ, биодоступность — 95–100 %, t1/2 — 5–7 часов. Показания к применению значительно расширены, например, инфекции нижних дыхательных путей (НДП) (обострение хронического бронхита, нозокомиальная пневмония); инфекции МВП; простатит; интраабдоминальные и тазовые инфекции (в сочетании с антианаэробными препаратами); кишечные инфекции (шигиллез, сальмонеллез); тяжелые инфекции кожи, мягких тканей, костей, суставов; гонорея; туберкулез (препарат II ряда); сибирская язва (лечение и профилактика).
Несколько уступает по активности ципрофлоксацину и офлоксацину пефлоксацин (абактал). Препарат почти на 100 % всасывается в ЖКТ. Всасывание фторхинолонов в ЖКТ резко ухудшается при одновременном приеме антацидов, сукральфата, препаратов, содержащих катионы Са, Mg, Al, Fe, Zn. Лучше других фторхинолонов пефлоксацин проникает через гематоэнцефалический барьер, t1/2 — 9–13 часов. Чаще других фторхинолонов может вызывать тендиниты. Показания к применению такие же, как и у офлоксацина, плюс вторичный бактериальный менингит в нейрохирургии.
Меньшей антимикробной активностью, чем другие фторхинолоны, особенно в отношении пневмококков, обладает ломефлоксацин (максаквин). Препарат не действует на P.aeruginosa. Имеет высокую биодоступность при приеме внутрь (около 100 %), t1/2 — 7–8 часов. Переносится несколько хуже, чем другие фторхинолоны. В частности, чаще вызывает фотосенсибилизацию.
Показания к применению ограничены инфекциями НДП (обострение хронического бронхита непневмококковой этиологии); инфекциями МВП. В России препарат применяется в комплексной терапии туберкулеза, однако контролируемые клинические исследования не проводились.
В целом препараты этой группы имеют целый ряд значительных преимуществ по сравнению с хинолонами I поколения.
По фармакодинамике — более широкий спектр активности, включающий:
— стафилококки (в том числе PRSA);
— грамотрицательные кокки (гонококк, менингококк, M.catarrhalis);
— грамположительные палочки (листерии, коринебактерии, возбудители сибирской язвы);
— грамотрицательные палочки семейства Enterobacteriaceae, включая полирезистентные (E.coli, сальмонеллы, шигеллы, протеи, энтеробактеры, клебсиеллы, серрации, провиденции, цитробактеры, морганеллы), P.aeruginosa, а также кампилобактеры;
— отдельные препараты (ципрофлоксацин, офлоксацин, ломефлоксацин и др.) активны против М.tuberculosis;
— действуют на некоторые внутриклеточные микроорганизмы (легионеллы).
По фармакокинетике:
— создают высокие концентрации в крови и тканях при приеме внутрь, биодоступность не зависит от времени приема пищи;
— хорошо проникают в различные органы и ткани: легкие, почки, простату;
— имеют длительный t1/2, назначаются 1–2 раза в день по переносимости;
— нежелательные реакции со стороны ЖКТ и ЦНС встречаются реже;
— могут быть использованы при почечной недостаточности.
Микробиологические особенности хинолонов II поколения не всегда могут устроить практикующего врача:
— малочувствительны к ним большинство стрептококков (в том числе пневмококки), энтерококки, хламидии, микоплазмы;
— не действуют на спирохеты, листерии и большинство анаэробов.
Итак, антистрептококковая активность препаратов группы ципрофлоксацина/офлоксацина явно невелика. Другим недостатком этой группы лекарственных средств является то, что они не имеют значения в лечении инфекций дыхательных путей, при которых возбудителями часто являются также пневмококки. И вот здесь проходит принципиальный «водораздел», объясняющий, почему появились новые фторхинолоновые препараты, которые получили и другое, почти официальное название — антипневмококковые фторхинолоны. В отличие от более ранних фторхинолонов данные препараты проявляют и антистафилококковую и антистрептококковую активность, к тому же они действуют на энтерококки. Антипневмококковые фторхинолоны действуют на хламидии и микоплазмы, а также анаэробы. Однако в качестве специфических антианаэробных препаратов их использование нерационально. Таким образом, по спектру действия новые фторхинолоны приближаются к карбапенемам, и если учесть, что все фторированные хинолоны очень хорошо проникают внутрь клеток, то понятно, какое мощное оружие получила медицина в лице этих препаратов.
Нет никакого сомнения в том, что новые фторхинолоны, то есть III–IV поколения, преодолели многие недостатки старых фторхинолонов. Одним из первых и основных препаратов III поколения оказался левофлоксацин (таваник), активность которого против пневмококков (включая пенициллинрезистентные штаммы) и атипичных возбудителей превосходила предыдущие классические фторхинолоны. Повышенная антипневмококковая активность левофлоксацина позволила Комиссии по контролю качества продуктов питания и лекарственных средств США одобрить его применение при внебольничных пневмониях, вызванных пенициллинрезистентными пневмококками. Левофлоксацин стал первым из фторхинолонов, получивших такое разрешение. Левофлоксацин — оптически активный левовращающий изомер офлоксацина — L-офлоксацин. Левофлоксацин характеризуется широким спектром действия, аналогичным офлоксацину, однако превосходит его по активности в отношении грамположительных кокков. То есть он превосходит своих предшественников в отношении стрептококков, ряда представителей энтеробактерий. По сравнению с офлоксацином обладает лучшим фармакологическим профилем, меньшей частотой развития побочных реакций и лучшей переносимостью. Левофлоксацин активнее офлоксацина в отношении Haemophilus influenzae, Moraxella catarralis, Mycoplasma pneumoniae, а в отношении Chlamydia pneumoniae превосходит ципрофлоксацин (табл. 3).
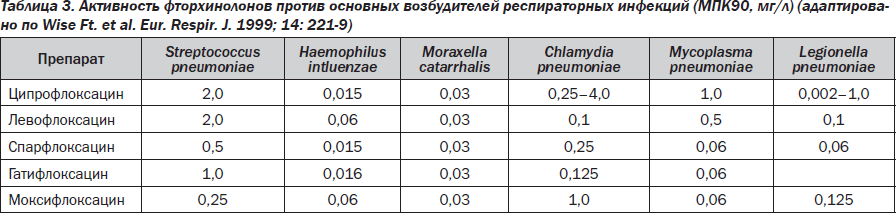
По результатам исследования PROTEKT (2001), левофлоксацин не уступает, а в ряде случаев превосходит по своей активности пенициллин, цефуроксим, кларитромицин, амоксициллин/клавуланат, которые традиционно считаются эффективными при заболеваниях, вызванных S.pneumoniae, М.catarralis, H.influenzae. Препарат имеет высокую биодоступность при приеме внутрь — около 100 %, t1/2 — 6–8 часов. Левофлоксацин хорошо проникает в воспалительный экссудат кожного волдыря у человека: после однократного введения 500 мг средние концентрации в экссудате (3 мг/л) достигались через 3,7 часа, а показатель проникновения в воспалительную жидкость — от 83 до 112 %.
Отмечается хорошее проникновение левофлоксацина в ткани дыхательных путей: после приема 500 мг средняя максимальная концентрация в легочной ткани составила 11,3 мг/кг; концентрация препарата в легких была в 2,5–5 раз выше, чем в плазме, и в течение 24 ч значительно превышала минимальную подавляющую концентрацию (МПК) для респираторных патогенов (S.pneumoniae, С.pneumoniae, M.catarralis, H.influenzae). Высокие концентрации левофлоксацина обнаруживались в слизистой бронхов (показатель проникновения составлял 90–180 % и жидкости эпителиальной выстилки — 8–300 %).
Показания к применению левофлоксацина — инфекции верхних дыхательных путей (ВДП) (острый синусит), инфекции НДП (обострение хронического бронхита, внебольничная пневмония), инфекции МВП, инфекции кожи и мягких тканей, сибирская язва (лечение и профилактика).
Левофлоксацин хорошо проникает и накапливается в больших количествах в клетках макроорганизма: в нейтрофилах, лимфоцитах, макрофагах. Соотношение внутриклеточных концентраций левофлоксацина в нейтрофилах к внеклеточным концентрациям составляло 8,8, а в полиморфонуклеоцитах — 9,8.
В альвеолярных макрофагах концентрации препаратов превышают сывороточные в 6 раз. Высокие концентрации левофлоксацина в клетках макроорганизма имеют большое значение для лечения инфекций с внутриклеточной локализацией возбудителей. Дальнейшие модификации химической структуры привели к появлению соединений, активных и в отношении анаэробов. Однако многие из вновь разработанных препаратов не достигли больных, так как были быстро отозваны с рынка вследствие развития тяжелых реакций. Генеалогическая схема развития хинолонов/фторхинолонов изображена на рис. 2.
«Все во имя человека, все для блага человека». Одним из новых препаратов, который стал успешно применяться в клинике, оказался моксифлоксацин (авелокс), представитель IV поколения фторхинолонов.
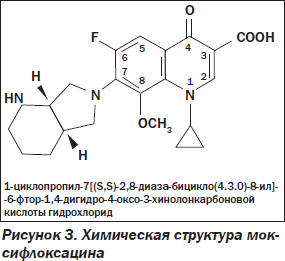 Наиболее важными структурами в молекуле фторхинолонов вообще и моксифлоксацина в частности, отвечающими за антимикробные свойства, являются группы, занимающие позиции 1, 7 и 8. Циклопропиловая группа в положении 1 обеспечивает активность против грамотрицательных микроорганизмов (рис. 3). Присоединение дополнительного кольца в позиции 7 придает высокую активность по отношению к грамположительной микрофлоре, включая пневмококки. Добавление в структуру молекулы метоксигруппы в положении 8 привело к повышению активности в отношении анаэробов без увеличения риска потенциальной фототоксичности.
Наиболее важными структурами в молекуле фторхинолонов вообще и моксифлоксацина в частности, отвечающими за антимикробные свойства, являются группы, занимающие позиции 1, 7 и 8. Циклопропиловая группа в положении 1 обеспечивает активность против грамотрицательных микроорганизмов (рис. 3). Присоединение дополнительного кольца в позиции 7 придает высокую активность по отношению к грамположительной микрофлоре, включая пневмококки. Добавление в структуру молекулы метоксигруппы в положении 8 привело к повышению активности в отношении анаэробов без увеличения риска потенциальной фототоксичности.
Моксифлоксацин (авелокс) превосходит хинолоны II поколения по активности против пневмококков (включая штаммы, устойчивые к пенициллину и макролидам) и атипичных патогенов (хламидии, микоплазмы).
В отличие от других фторхинолонов моксифлоксацин хорошо действует на неспорообразующие анаэробы, в том числе B.fragilis. Несколько уступает ципрофлоксацину по активности в отношении грамотрицательных бактерий семейства Enterobacteriaceae и синегнойной палочки. Биодоступность при приеме внутрь — 90 %, t1/2 — 12–13 часов.
Показания к применению касаются инфекций ВДП (острый синусит), инфекций НДП (обострение хронического бронхита, внебольничная пневмония), инфекции кожи и мягких тканей.
Как и все фторхинолоны, моксифлоксацин действует бактерицидно благодаря блокированию синтеза бактериальной ДНК, ингибируя ферменты класса топоизомераз — ДНК-гиразы (топоизомеразы II) и топоизомеразы IV (рис. 4).
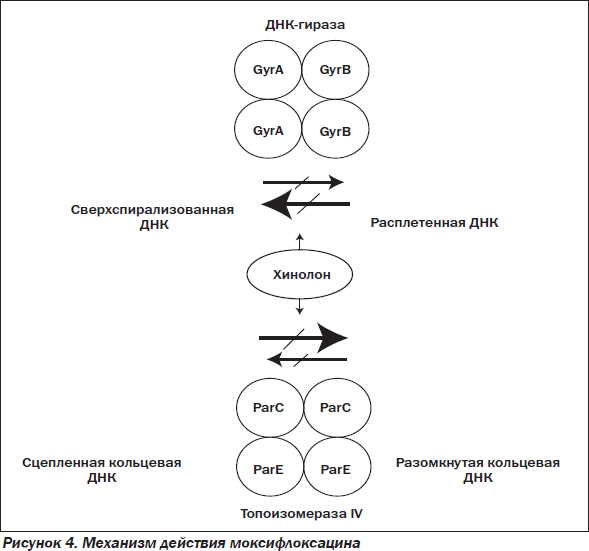
Топоизомераза IV — это еще один фермент бактериальной ДНК (после ДНК-гиразы) и вторая мишень для фторхинолонов, работает координировано с ДНК-гиразой, принимая участие в общем процессе репликации ДНК. Топоизомераза IV катализирует декатинацию — расщепление двух связанных нитей ДНК после репликации, то есть отделение дочерних молекул ДНК. ДНК-гираза работает впереди репликативной вилки, удаляя избыток позитивных супервитков, топоизомераза IV — позади.
Итак, эти ферменты выполняют строго определенные функции в процессе формирования пространственной структуры молекулы ДНК при ее репликации: ДНК-гираза катализирует расплетение (отрицательную суперспирализацию) нитей ДНК, а топоизомераза IV участвует в разъединении (декатинации) ковалентно-замкнутых кольцевых молекул ДНК. Ингибирование этих ферментов нарушает процессы роста и деления бактериальной клетки, что приводит к ее гибели.
Классические фторхинолоны действуют только на один фермент, в то время как второй не ингибируется. Установлено, что главной мишенью у грамположительных микроорганизмов является топоизомераза IV, а у грамотрицательных патогенов — ДНК-гираза. Новые фторхинолоны пагубно влияют на оба фермента, вследствие чего значительно расширяется спектр их действия.
Одновременное влияние на два фермента существенно снижает вероятность появления резистентных штаммов микроорганизмов, так как чем больше активность препарата в отношении обоих ферментов, тем ниже уровень резистентности, обусловленной мутацией в генах, кодирующих один фермент.
В целом группа фторхинолоновых антибиотиков отличается хорошими, а ряд из них — отличными фармакокинетическими показателями. В первую очередь обращает на себя внимание высокая биодоступность этой группы антибиотиков. Причем если у офлоксацина и пефлоксацина она колеблется в пределах 95–100 %, то у левофлоксацина она стабильно держится на уровне 100 %.
Максимальная концентрация в крови фторхинолонов колеблется от 1,2 до 5,3, причем у новых фторхинолонов она достигает показателя 5,0 и выше.
Прогнозировать эффективность дозозависимого антибиотика можно с помощью еще одного фармакодинамического параметра — отношение площади под фармакологической кривой (AUC) к МПК или к МБК. AUC отражает изложение концентрации препарата в крови от момента его введения до полной элиминации из организма или в течение 24 часов после введения. Чем выше AUC/МПК, тем эффективнее препарат. Установлено, что уничтожение чувствительных микроорганизмов достигается при величине AUC/МПК не меньше 30. У левофлоксацина AUC/МПК составляет 50, то есть почти в 1,7 раза превышает необходимую величину для пневмококков.
Фторхинолоны способны подавлять размножение микроорганизмов в концентрациях, ниже МПК, — так называемый субМПК эффект. Они также обладают выраженным постантибиотическим эффектом (ПАЭ) против грамположительных и грамотрицательных микроорганизмов, который в среднем равен 2.
Напомним, что антибактериальные препараты дозируются с тем расчетом, чтобы их концентрации в очаге воспаления превышали МПК того или иного патогена. Однако после уменьшения концентрации антибиотика ниже МПК рост и размножение бактерий начинаются не сразу. Период отсутствия их жизнедеятельности определяется как ПАЭ. Чрезвычайно важным является достоверное доказательство этого явления у респираторных фторхинолонов. ПАЭ доказано у левофлоксацина для таких патогенов, как S.pneumoniae, S.aureus, E.coli, S.epidermis, B.fragitis.
ПАЭ моксифлоксацина для разных микроорганизмов составляет 1,2–3,1 ч при концентрации, равной 4+МПК, и увеличивается с возрастанием концентрации препарата. Например, ПАЭ для S.pneumoniae равно 2,2 ч при концентрации препарата 4+МПК и возрастает до 2,7 при его концентрации 10+МПК.
Таким образом, фторхинолоны III–IV поколений открыли новые перспективы в лечении инфекций различных локализаций. В частности, в современных руководствах по лечению внебольничной пневмонии наряду с b-лактамами и макролидами рекомендуются новые фторхинолоны, особенно в регионах, где появились полирезистентные пневмококки.
Преимущества фторхинолонов заключаются в возможности перорального и парентерального применения, высокой активности и хорошей переносимости, низком риске развития устойчивых штаммов. Однако чрезвычайно широкое применение фторхинолонов в животноводстве рисует клиническую бесперспективность этой группы препаратов. За время клинического изучения и широкого применения фторхинолонов в медицинской практике накоплены данные, показывающие возможное нарастание частоты выделения клинических штаммов бактерий с устойчивостью к фторхинолонам. Развитию резистентности способствуют и длительные курсы лечения. Частота спонтанных мутаций к фторхинолонам очень низкая. Возникновение приобретенной резистентности у бактерий к фторхинолонам связано в первую очередь с изменением свойств (чувствительности) двух основных ферментов-мишеней: снижением чувствительности к фторхинолонам субъединиц ДНК-гиразы и топоизомеразы IV. Это зависит от соответствующих мутаций в генах, кодирующих эти ферменты.
Причиной развития резистентности к фторхинолонам может быть нарушение транспортных систем клетки. Это связано с повреждением системы пориновых каналов, образуемых пориновым белком OmpF. Соответственно снижается степень пассивной диффузии в первую очередь гидрофильных фторхинолонов (ципрофлоксацин). Возможно также изменение структуры липополисахаридного слоя мембраны бактериальной клетки и снижение проникновения в клетку липофильных фторхинолонов (офлоксацина). Через пориновые каналы в бактериальную клетку проникают также b-лактамы, тетрациклины, хлорамфеникол, аминогликозиды и некоторые другие антибактериальные препараты. Поэтому при нарушении этого транспортного пути может развиваться перекрестная устойчивость одновременно к фторхинолонам и химиотерапевтическим препаратам других структурных классов. Следует иметь в виду и возможную высокую устойчивость к фторхинолонам метициллинрезистентных штаммов стафилококков. Например, в госпиталях Австралии выделяли от 16–24 до 80–100 % устойчивых к фторхинолонам штаммов среди метициллинрезистентных стафилококков. Поистине будущее хинолонов зависит от будущего резистентности. Тем не менее дальнейшие изменения структуры фторхинолонов могут привести к созданию нерезистентных препаратов более широкого или узконаправленного действия.