
Газета «Новости медицины и фармации» 9 (415) 2012
Вернуться к номеру
Новости МЗ
Українці отримали законне право повідомляти про побічні реакції при застосуванні ліків
Наказом Міністерства охорони здоров’я України від 27.12.2006 р. № 898, зареєстрованим в Міністерстві юстиції України 29.01.2007 р. № 73/13340, зі змінами, внесеними наказом МОЗ України від 29.12.2011 р. № 1005 (зареєстровано в Міністерстві юстиції України 15.03.2012 р. № 402/20715), затверджено Порядок здійснення нагляду за побічними реакціями лікарських засобів, дозволених до медичного застосування.
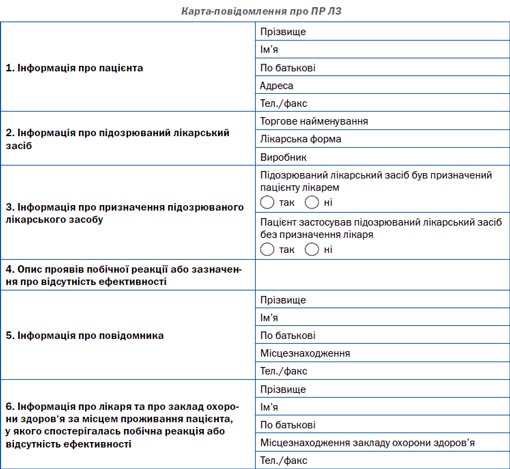
На сьогодні в Україні законодавчо передбачено, що пацієнт, його представник або організація, яка представляє або захищає інтереси пацієнта, можуть повідомити про побічну реакцію та/або відсутність ефективності лікарського засобу при його медичному застосуванні. У додатку 3 до наказу № 898 міститься карта-повідомлення про побічну реакцію та/або відсутність ефективності лікарського засобу та правила її заповнення.
Електронна форма карти-повідомлення розміщена на сайті ДП «Державний експертний центр МОЗ України» (www.pharma-center.kiev.ua) (розділ «Фармаконагляд», підрозділ «Карта-повідомлення про ПР ЛЗ», у якому знаходиться форма карти-повідомлення, що заповнюється пацієнтом), а також на сайті Всеукраїнської ради захисту прав та безпеки пацієнтів (www.medrada.org).
Карта-повідомлення для надання пацієнтом та/або його представником, організаціями, які представляють інтереси пацієнтів, інформації про побічну реакцію та/або відсутність ефективності лікарського засобу при його медичному застосуванні або відсутність ефективності заповнюється та подається за адресою: ДП «Державний експертний центр МОЗ України», Управління післяреєстраційного нагляду, вул. Ушинського, 40, м. Київ, 03151; тел./факс: +38 (044) 498-43-58; e-mail: bezpecapacienta@pharma-center.kiev.ua; електронна форма повідомлення розміщена на www.pharma-center.kiev.ua.
Кабмін виключив препарати амітриптилін і левана зі списку наркотичних речовин
Кабінет Міністрів України вніс зміни до переліку наркотичних засобів, психотропних речовин і прекурсорів, який затверджено постановою Кабінету Міністрів України від 6 травня 2000 року № 770. Відповідну постанову прийнято 23 травня 2012 року. Це рішення продиктоване піклуванням уряду про здоров’я нації і зумовлене пріоритетністю медичного складника в заходах контролю за обігом психоактивних речовин, повідомили у прес-службі Державної служби з контролю за наркотиками.
Державна служба з контролю за наркотиками, визнаючи те, що використання відповідних речовин для медичних і наукових цілей не повинне надмірно зарегульовуватися, опрацювала із залученням науковців, представників правоохоронних органів та громадських організацій питання доцільності обмеження обігу амітриптиліну й левани та внесення до зазначеного переліку деяких рослин, що в природних умовах в Україні не ростуть, а культивуються лише в обмежених кількостях для наукових цілей.
Проведені клінічні дослідження показали, що амітриптилін і левана є ефективними й доступними препаратами для лікування депресивних розладів психіки. Водночас, за даними правоохоронних органів, не зафіксовано фактів немедичного вживання згаданих ліків наркозалежними особами. Корективи, внесені до переліку, є цілком виправданими й узгодженими з міжнародними договорами, тож реалізація постанови Кабінету Міністрів України поліпшить доступ до лікарських препаратів для надання психіатричної допомоги, усуне дублювання та внесе ясність у положення нормативних актів Уряду щодо контролю за психоактивними компонентами та рослинами, які їх містять.
МОЗ роз’яснило нормативне регулювання
етичних та морально-правових аспектів клінічних випробувань
До Державного експертного центру МОЗ надійшов лист-роз’яснення Департаменту правового забезпечення та міжнародної діяльності МОЗ України щодо нормативного регулювання етичних та морально-правових аспектів клінічних випробувань лікарських засобів в Україні.
У листі зазначено, що нормами Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань, затвердженого наказом МОЗ України від 23.09.09 № 690, було передбачено обо-в’язкове отримання схвалення Центральної комісії з питань етики МОЗ України до початку клінічного випробування лікарського засобу, його проведення, закінчення та внесення суттєвих виправлень до матеріалів такого випробування тощо.
Такі положення суперечили вимогам частини 7 статті 7 Закону України «Про лікарські засоби», згідно з якими клінічні випробування лікарських засобів проводять після обов’язкової оцінки етичних та морально-правових аспектів програми клінічних випробувань комісіями з питань етики, які створюються і діють при лікувально-профілактичних закладах, де проводять клінічні випробування. З метою приведення процеду-ри оцінки етичних та морально-правових аспектів програми клінічних випробувань лікарських засобів у відповідність до статті 7 Закону України «Про лікарські засоби» наказом МОЗ України від 11.04.12 № 255 «Про упорядкування етичних аспектів клінічних випробувань лікарських засобів» (надалі за текстом — наказ № 255) було визнано таким, що втратив чинність, наказ МОЗ України від 17.07.06 № 485 «Про утворення та склад Центральної комісії з питань етики Міністерства охорони здоров’я України».
Пунктом 2 наказу № 255 Управлінню розвитку фармацевтичного сектора галузі охорони здоров’я МОЗ України доручено у двомісячний строк внести зміни до Порядку проведення клінічних випробувань лікарських засобів та експертизи матеріалів клінічних випробувань, затвердженого наказом МОЗ України від 23.09.09 № 690.
Таким чином, наразі на підставі чинного законодавства схвалено початок клінічного випробування лікарського засобу та внесення суттєвих поправок до його матеріалів, а також те, що нагляд за проведенням таких випробувань та їх закінченням має здійснюватись комісіями з питань етики, які створюються і діють при лікувально-профілактичних закладах, у яких проводять такі клінічні випробування.