
Журнал «Здоровье ребенка» 5 (56) 2014
Вернуться к номеру
Аутовоспалительные синдромы в практике врача-педиатра
Авторы: Недельская С.Н., Жиленко И.А., Мережко А.С., Шапран А.В. - Запорожский государственный медицинский университет
Рубрики: Педиатрия/Неонатология
Разделы: Справочник специалиста
Версия для печати
В обзорной статье предоставлены информация, характеристика, определение и классификация заболеваний, которые были отнесены к отдельной нозологической группе и названы аутовоспалительными заболеваниями или синдромами (АВС). Концепция АВС сформирована в конце 1990-х; изучение и накопление данных активно продолжаются в настоящее время, и с этой целью был создан международный регистр АВС в рамках программы EUROFEVER. Дана краткая характеристика как отдельных групп заболеваний с типичными генетическими мутациями, так и некоторых нозологических форм. Перечислены препараты с доказанной терапевтической эффективностью, в том числе НПВП, ГКС и генно-инженерные биологические препараты.
В оглядовій статті надані інформація, характеристика, визначення та класифікація захворювань, що були віднесені в окрему нозологічну групу та названі автозапальними захворюваннями або синдромами (АЗС). Концепцію АЗС сформовано наприкінці 1990-х рр.; вивчення та накопичення даних активно триває і зараз, і з цією метою був створений міжнародний регістр АЗС у рамках програми EUROFEVER. Подано коротку характеристику як окремих груп захворювань із типовими генетичними мутаціями, так і деяких нозологічних форм. Перераховано препарати з доведеною терапевтичною ефективністю, у тому числі НПЗП, ГКС та генно-інженерні біологічні препарати.
This review article provides information, characterization, identification and classification of diseases that have been attributed to a separate nosological group and called autoinflammatory diseases or syndromes (ABC). ABC concept was formed in the late 1990s; study and data accumulation are still actively pursued at the moment, and for this purpose there has been created international ABC register as a part of EUROFEVER program. Brief characteristics of both individual groups of diseases with typical genetic mutations and some clinical entities are provided. Drugs with proven therapeutic efficacy, including NSAIDs, GCs and genetically engineered biological agents, are listed.
аутовоспалительные синдромы, семейная средиземноморская лихорадка, криопирин-ассоциированные синдромы, гипер-IgD-синдром.
автозапальні синдроми, сімейна середземноморська лихоманка, кріопірин-асоційовані синдроми, гіпер-IgD-синдром.
autoinflammatory syndromes, familial Mediterranean fever, cryopyrin-associated syndromes, hyper-IgD-syndrome.
Статья опубликована на с. 66-72
В последние десятилетия все более пристальное внимание специалистов привлекают заболевания, которые составили группу аутовоспалительных заболеваний или синдромов (АВС) (Human autoinflammatory diseases or syndromes — HAIDS). Согласно определению P. Fietta (2004), ABC/HAIDS — это гетерогенная группа генетически детерминированных состояний, характеризующаяся не связанными с инфекционной контаминацией приступами воспаления и манифестирующая лихорадкой и клинической симптоматикой, типичной для ревматических или аутоиммунных заболеваний, при отсутствии аутоиммунных или инфекционных причин. Чаще всего эти заболевания дебютируют в детском возрасте, поэтому врачи-педиатры, детские ревматологи будут первыми сталкиваться с этими достаточно сложными для диагностики случаями [7].
Концепция АВС созрела сравнительно недавно, в конце 1990-х, вследствие накопившихся данных о различных гетерогенных заболеваниях, протекающих с разными клиническими симптомами, характеризующимися прогрессирующим или интермиттирующим течением с проявлениями системного воспаления, связанного с генетически детерминированной гиперпродукцией интерлейкинов и провоспалительных факторов.
В настоящее время уже создана классификация болезней и синдромов HAIDS, однако она постоянно расширяется и пополняется новыми заболеваниями с учетом новых полученных данных. В 2002 году был создан проект EUROFEVER в рамках педиатрического европейского ревматологического сообщества (PRES). В рамках этого проекта создан регистр больных, в котором уже зарегистрированы 1880 пациентов из 31 страны, у 95 % из них диагнозы были подтверждены генетически. Составлен перечень заболеваний согласно проекту EUROFEVER, которые отнесены к группе аутовоспалительных синдромов [25].
Перечень заболеваний согласно регистру EUROFEVER:
— синдром Бехчета;
— синдром Блау, или раннее начало саркоидоза;
— криопирин-ассоциированные периодические синдромы;
— хронический рецидивирующий мультифокальный остеомиелит;
— болезни, обусловленные недостаточностью антагониста рецептора IL-1;
— семейная средиземноморская лихорадка;
— недостаточность мевалонаткиназы (гипер-IgD-синдром);
— NRLP-12 ассоциированный периодический синдром;
— пиогенный стерильный артрит, пиодермия гангренозная и акне (PAPA-синдром);
— периодический синдром, ассоциированный с рецепторами TNF-а (TRAPS);
— периодическая лихорадка, афтозный стоматит, фарингит и шейный аденит (PFAPA);
— недифференцированная периодическая лихорадка;
— DIRA/DITRA синдром;
— синдром Шницлера;
— Маджед-синдром.
Большая часть перечисленных синдромов и болезней относятся к редким заболеваниям с первыми проявлениями в раннем детском возрасте и достаточно трудны для диагностики. Общий и ведущий симптом этих заболеваний — это длительная или периодическая лихорадка, которая требует мультидисциплинарного дифференциально-диагностического подхода с исключением хронических или рецидивирующих инфекций, ревматических болезней и аутоиммунных состояний. Из других наиболее часто встречающихся симптомов можно назвать:
— мышечно-артикулярный;
— кожный синдром (разнообразные кожные сыпи);
— воспаление серозных оболочек;
— возможное развитие амилоидоза;
— высокие лабораторные острофазовые маркеры воспаления (СРБ, СОЭ, лейкоцитоз);
— отсутствие аутоантител или активации аутоспецифических клеток.
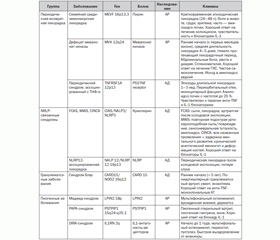
Основное звено патогенеза всех групп и синдромов АВС — это гиперактивация антигенспецифического иммунитета и гиперпродукция острофазовых реактантов — СРБ, серомукоидов, СОЭ, сывороточного амилоида А, лейкоцитоз и др. Основные медиаторы воспаления — IL-1, TNF-а и др. Причиной большинства синдромов является мутация одного гена или генов-модификаторов, вследствие чего может изменяться течение заболевания (табл. 1).
/68/68.jpg)
Впервые описание пациентов со средиземноморской лихорадкой было сделано в 1908 году Janeway and Mosenthal. Однако активное изучение этого заболевания началось только в 1948 г., когда Reiman опубликовал описание нескольких пациентов с периодической лихорадкой с дебютом заболевания в детском возрасте и персистированием на протяжении нескольких лет или десятилетий и назвал это заболевание периодической болезнью. Углубленное изучение этих заболеваний началось в конце ХХ века с появлением возможности проведения генетических исследований и расшифровкой генетических мутаций. Так, в 1982 г. был описан TRAPS-синдром, в 1984 г. — гипер-IgD-синдром — HIDS, синдром периодической лихорадки, ассоциированный с рецепторами к фактору некроза опухоли, — TRAPS-синдром, синдром Макла — Уэльса — MWS и многие другие. Список заболеваний постоянно пополняется, а кроме всего прочего, пересматривается патогенез многих из них, и сейчас такие болезни, как субсепсис Висслера — Фанкони, системная форма ЮРА, подагра, семейный хондрокальциноз (псевдоподагра), саркоидоз, генерализованные формы псориаза, болезнь Гоше, идиопатический фиброзирующий альвеолит, болезнь Бехчета и др., также рассматриваются как системные аутовоспалительные заболевания.
Семейная средиземноморская лихорадка (Familial Mediterranian Fever — FMF)
FMF — первый из описанных и наиболее часто встречающихся АВС. Этому заболеванию подвержены в основном 4 этнические группы: евреи, арабы, турки, армяне [6, 18, 32, 33]. Дебют заболевания у большинства пациентов относится к возрасту до 20 лет. Его основные клинические проявления: эпизоды лихорадки до 40 °С и выше, которые продолжаются от 6 ч до 3–4 суток; атаки гипертермии с периодичностью в 3–4 недели; часто сопровождается полисерозитом — абдоминальным синдромом вследствие асептического перитонита, асептическим плевритом, перикардитом; может наблюдаться артрит, чаще моноартрит (гонит) с большим количеством выпота; возможны эризипелоидподобная сыпь на голенях и стопах, отек и болезненность мошонки, гепатоспленомегалия, разнообразные неврологические и психоневрологические синдромы. Во время атаки обычно повышены СОЭ, СРБ, сывороточный амилоид А (SAA), лейкоцитоз с нейтрофилезом. Осложнение данного заболевания — часто развивающийся амилоидоз почек или ЖКТ, нередко приводящий к гибели больных. Для диагностики этого заболевания используют критерии Тель-Хашомера. Проводится молекулярно-генетическое типирование гена MEFV. В базисном лечении этого заболевания с 1972 г. используют колхицин, что коренным образом изменило прогноз у пациентов с FMF. Максимальная доза колхицина для пациентов — 2 мг/сут. Даже если колхицин не предотвращает рецидив заболевания, он значительно снижает риск развития амилоидоза. К базисному препарату можно добавить симптоматическое лечение нестероидными противовоспалительными препаратами (НПВП). В настоящее время при резистентности к колхицину можно усилить терапию генно-инженерными биологическими препаратами (ГИБП): блокаторами IL-1 (анакинра) и блокаторами TNF-а (инфликсимаб).
Гипер-IgD-синдром — синдром дефицита мевалонат киназы (Hyper IgD-syndrome — Mevolonate-Kinase Deficiency-syndrome — HIDS/MVKD)
Данный синдром был описан как самостоятельное заболевание в 1984 г. J.W.M. van der Meer. Тип наследования — аутосомно-рецессивный, обусловлен мутацией гена MVK, расположенного на длинном плече 12-й хромосомы. Чаще встречается у датчан, голландцев, французов.
Заболевание обычно начинается в раннем детском возрасте, возможно на первом году жизни. Первый приступ может быть спровоцирован травмой, хирургическим вмешательством, вакцинацией и т.д. Клинические проявления заболевания: рецидивирующая лихорадка с ознобами, с длительностью пирексии от 2 до 7 дней и периодичностью в 2–8 недель; пятнисто-папулезная, уртикарная или петехиальная сыпь; шейная лимфаденопатия с болезненными лимфоузлами; абдоминальный синдром (боль, рвота, диарея), гепатоспленомегалия; артралгии или артриты крупных суставов (недеструктивные); оральные и/или генитальные язвы, офтальмологические симптомы. В периоде обострения повышены острофазовые показатели (лейкоцитоз, СРБ, SAA). Диагностические критерии заболевания — IgD выше 100 МЕ/мл, а также возможное повышение уровня IgA. Подтверждает диагноз выявление мутации гена MVK. Лечение данного синдрома остается проблематичным. Такие препараты, как колхицин и иммуносупрессоры, оказались малоэффективными. Более обнадеживающие результаты получены от использования ГИБП — анакинры или этанерцепта. В качестве симптоматических препаратов используют НПВП. Однако, несмотря на остроту приступов, прогноз заболевания благоприятный, так как амилоидоз развивается редко. Полное выздоровление маловероятно [2, 3, 6, 10].
NRLP, или криопирин-ассоциированные синдромы
Общей генетической основой этих синдромов является мутация гена CIAS1, расположенного на длинном плече 1-й пары хромосом. Данный белок является основой супермолекулярного комплекса, обеспечивающего активную форму ИЛ-1b, а также выполняющего программу апоптоза.
Клиника семейного холодового аутовоспалительного синдрома/семейной холодовой крапивницы (Familial Cold Autoinflammatory Syndrome/Familial Cold Urticaria — FCAS/FCU): лихорадка, артралгии/артрит, миалгии, уртикарная сыпь, конъюнктивит, эписклерит, иридоциклит, высокая активность острофазовых показателей, чаще инициированные экспозицией холода. Симптомы возникают через 1 час после воздействия холода, пик симптомов — через 6–8 часов, исчезают симптомы через 24 часа. Амилоидоз развивается редко. Лечение: необходимо избегать воздействия холода; симптоматически назначаются НПВП; базис-терапия — высокие дозы глюкокортикостероидов (ГКС); терапия ингибиторами ИЛ-1 — анакинрой, риланосептом продемонстрировала высокую эффективность [5, 9, 11, 12].
Синдром Макла — Уэльса (Muckle — Wells syndrome — MWS) по тяжести занимает промежуточное положение среди криопирин–ассоциированных синдромов. Он характеризуется повторными эпизодами лихорадки и сыпи, длящимися 24–48 часов, артритами крупных суставов, поражением глаз, в 50–70 % случаев — развитием нейросенсорной глухоты, обычно у подростков или взрослых. Амилоидоз — частое осложнение синдрома Макла — Уэльса. Лечение: ГКС в высоких дозах, ГИБП — этанерцепт, анакинра [13, 14, 16, 17].
Синдром CINCA-NOMID (Сhronic Infantile Neurological Cutaneus Arthicular). Путь наследования — аутосомно-доминантный, ген CIAS1, кодирующий белок криопирин. Среди криопирин-ассоциированных заболеваний синдром CINCA-NOMID протекает клинически наиболее тяжело, с ранним дебютом и плохим прогнозом для пациентов. Клинические проявления начинаются на первом году жизни с эпизодов лихорадки или непрерывной лихорадки, с персистирующей уртикарной сыпью, симметричной артропатией крупных суставов, с эпифизарными и метафизарными изменениями, развитием контрактур, частыми глазными симптомами: прогрессирующими увеитами, отеком, атрофией зрительного нерва, вплоть до развития слепоты. Обычно развивается задержка умственного развития, возможны асептический менингит, гидроцефалия, атрофия коры головного мозга, развитие нейросенсорной глухоты. Эти клинические проявления сопровождаются высокой активностью острофазовых показателей. Амилоидоз возможен в 20 % случаев. Смертность высокая. Лечение: ГКС в высоких дозах + ГИБП (анакинра п/к 1 мг/кг) [13, 16, 33].
Синдром Маршалла (Periodic Fever, Aphthous Stomatitis, Pharingitis, Cervical Adenitis — PFAPA)
Это заболевание редкое, с неизвестным типом наследования, неизвестен также конкретный мутантный ген. Цитокиновый профиль свидетельствует о повышении сывороточных уровней ИЛ-1b, ТНФ-а, ИЛ-6, ИЛ12р70 в периоды как обострения, так и ремиссии, что свидетельствует о постоянном субклиническом воспалении [3, 17, 31, 33].
Среди заболевших преобладают мальчики. Дебют заболевания чаще всего с 2 до 5 лет. Синдром PFAPA характеризуется периодичностью фебрильных приступов. Интервалы между атаками составляют от 2 до 8 недель и со временем удлиняются. Клинические критерии диагностики были разработаны Маршаллом: приступы лихорадки без признаков инфицирования, афтозный стоматит, шейный лимфаденит, тонзиллит/фарингит, лейкоцитоз, повышенная СОЭ. Отмечаются полное отсутствие симптомов между фебрильными приступами, нормальный рост и развитие ребенка. Лечение: НПВС и ГКС в высоких дозах; ГКС в дозе 1–2 мг/кг массы тела полностью прерывает приступ. Было отмечено, что тонзиллэктомия или аденотонзиллотомия часто приводит к полному выздоровлению ребенка [17].
Хронический рецидивирующий мультифокальный остеомиелит (CRMO, Maджед-синдром, PAPA-, DIRA-синдромы)
Синдром CRMO (Maджед-синдром) впервые был описан A. Gideon et al. в 1972 г. и K.H. Gustavson и H.F. Wilbrand в 1974 г. Причинный ген — LPIN2, кодирует липин-2 протеин. Клинические проявления: множественные очаги костной деструкции, при этом из очага деструкции невозможно выделить инфекционный фактор. Это заболевание детского возраста. Начало варьирует от 2 до 17 лет, возможен дебют и в более старшем возрасте. Cиндром СRMO может протекать как моноочаговое и многоочаговое заболевание, как с одним эпизодом обострения, так и с рецидивирующим течением. Кроме поражения костей, могут вовлекаться суставы и кожа. Сопровождается симптомами интоксикации, субфебрилитетом или фебрильной температурой, болями в пораженных сегментах скелета, часто достигающими высокой интенсивности. Чаще поражаются медиальный конец ключицы, лопатка, грудина, поясничный или грудной отдел позвоночника. Суставной синдром встречается в 80 % случаев, часто сопровождается энтезитами, но имеет транзиторный характер. Часто при CRMO наблюдается пустулез кожи с наиболее характерной локализацией на коже стоп и кистей (правило 3 «П»: пальмарно-плантарный пустулез). Как правило, при этом синдроме отмечается умеренное повышение активности острофазовых показателей (СРБ, СОЭ, лейкоцитоз). Часто ассоциировано с антигеном HLA-В27 и -В40.
Окончательный диагноз ставится на основании биопсии костного очага и исключения бактериального остеомиелита. Лечение: симптоматически НПВС, могут назначаться ГКС, бисфосфонаты, реже — препараты интерферона a или y. Также подтвердили свою эффективность ингибиторы ТNF — инфликсимаб, этанерцепт [31, 33].
PAPA-синдром (Pyogenic Arthritis, Pyoderma gangrenosum and Acne) — это семейная наследственная патология, передающаяся аутосомно-доминантным путем. Недавно был идентифицирован ген на 15–й хромосоме и выявлены 2 возможные мутации гена CD2BP1.
Клинические проявления РАРА-синдрома следующие. Заболевание дебютирует в детском или юношеском возрасте и стартует с поражения суставов. Течение артрита неблагоприятное, с развитием деструкции. Пиодермия не всегда сопровождает артрит или может развиться позже. Практически всегда у пациентов развивается акне, и если не проводится лечение, вызывает рубцовые поражения кожи. Лечение: наибольший эффект получен при назначении антагонистов ТНФ — этанерцепта, инфликсимаба или антагонистов ИЛ-1 — анакинры. Для лечения акне необходимы оральные тетрациклины.
DIRA-синдром (Deficiancy of the Il-1 receptor antagonist) — редкое генетически детерминированное заболевание, развивающееся у новорожденных. Описанные случаи выявлены у пуэрториканцев, ливанцев, голландцев. Путь передачи аутосомно-рецессивный, в гене IL1RN (2q14.2), находящемся во 2-й хромосоме. Клинические проявления наблюдаются в первые дни жизни новорожденного и представлены дистресс-синдромом, пустулезным поражением кожи, эрозиями на слизистой полости рта, отеком суставов и болезненностью их движений, гепатоспленомегалией. Из дополнительных признаков следует отметить генерализованный ихтиоз кожи, изменения ногтей по типу псориатического поражения, конъюнктивит, церебральный васкулит, гипотонию и прогрессирующую гипотрофию. Как правило, заболевание не сопровождается повышением температуры. При отсутствии адекватной терапии развивается полиорганная недостаточность, приводящая к летальному исходу. Для подтверждения диагноза необходимо исключить инфицирование, провести биопсию кожи (обнаруживают нейтрофильную инфильтрацию и тромбозы кожи), рентгенографию костей (для подтверждения баллоновидной деформации ребер, периостальной реакции длинных трубчатых костей, мультифокальных остеолитических повреждений костей), по показаниям — МРТ головного мозга для подтверждения васкулита. Лечение: при назначении анакинры достаточно быстро удается добиться регрессии заболевания и сохранить жизнь пациентам.
Болезнь Бехчета у детей
Болезнь Бехчета (ББ) в настоящее время рассматривается как системный васкулит неизвестной этиологии, характеризующийся язвенно–некротическими поражениями ротовой полости, гениталий, с частым поражением глаз, артритом, поражением ЖКТ, нервной системы и сосудов [8, 15, 19, 29, 30].
Чаще данная патология регистрировалась в Японии и странах Среднего Востока, а семейный характер поражений свидетельствует о возможных генетических мутациях. В настоящее время антиген HLA b5101 может рассматриваться как антиген риска развития ББ. Заболевание дебютирует в детском возрасте. Клиническая картина следующая. Заболевание чаще всего начинается с развития афт в полости рта, одиночных или множественных, имеющих рецидивирующее течение. Язвы гениталий появляются позже — в пубертатном возрасте. Поражения глаз развиваются у 60 % детей, преимущественно у мальчиков; часто выявляют двусторонний увеит, который при тяжелом течении способен привести к потере зрения. Может развиваться катаракта, глаукома. Поражения кожи наблюдаются у 90 % пациентов: узловатая эритема, пустулез, акне, фолликулит, пурпура, редко — язвы. У 15 % детей отмечаются неврологические поражения: церебральный васкулит, менингоэнцефалит, энцефалит. У 75 % детей имеет место артрит с вовлечением крупных суставов. Эрозивный и деструктивный процессы не характерны. Среди сосудистых проявлений чаще отмечаются артериальные или венозные тромбозы и аневризмы или окклюзии артерий в различных местах. Возможен амилоидоз внутренних органов.
Одним из критериев является тест развития патергии, характеризующийся образованием маленьких пустул или узелков в месте укола через сутки после инъекции. В стадии обострения имеет место нарастание острофазовых показателей, повышение уровня гаммаглобулинов. Терапия ББ: НПВС, ГКС, колхицин, иммуносупрессивная терапия — метатрексат, циклоспорин А. В последние годы была продемонстрирована эффективность использования ГИБП — инфликсимаба и этанерцепта [8, 15].
Недифференцированный аутовоспалительный синдром
Среди детей с АВС встречаются пациенты с длительно сохраняющейся или периодически рецидивирующей лихорадкой, артритами, повышением острофазовых показателей и другими признаками системного заболевания. Безусловно, эти больные требуют очень тщательного проведения дифференциально-диагностического анализа, исключения ряда инфекционных заболеваний (боррелиозов, ВИЧ, малярии, тифо-паратифозных заболеваний, лейшманиоза, туберкулеза и др.), онкогематологических заболеваний, ревматической патологии. Часто в начале заболевания отсутствует полный симптомокомплекс, что значительно затрудняет диагностику. Это всегда трудные в диагностическом плане пациенты, требующие мультидисциплинарного подхода и длительного катамнестического наблюдения. Поэтому было предложено введение термина «недифференцированный АВС» (кодируется по МКБ-10 как ЛНГ — лихорадка неясного генеза (780–6)) [33].
Вопрос о выборе терапии таким больным также должен решаться индивидуально. В ряде случаев при благоприятном течении заболевания вполне уместно не назначать терапию, проводя наблюдение за состоянием ребенка. В других случаях возможно проведение терапии ex juvantibus, при преобладании тех или иных синдромов и симптомов. АВС — это достаточно редкие заболевания, но знание этих синдромов клиницистам просто необходимо для своевременной диагностики. В последние годы стала возможна терапия генно-инженерными биологическими препаратами. Поэтому корректная и своевременная диагностика является залогом излечения, а иногда и спасения жизни пациента. Изучение данной патологии во всем мире набирает обороты, значительно вырос интерес к данной группе заболеваний. В связи с этим ежегодно описываются новые синдромы с генетической расшифровкой причин возникновения заболеваний, а также расширяются наши знания, углубляется понимание известных ранее заболеваний с новыми дополнениями к механизму их развития, а соответственно, и с новыми методами лечения.
1. Arostegui J.I., Solis P., Aldea A. et al. Etanercept plus colchicines treatment in a child with tumor necrosis factor receptor-associated periodic syndrome abolished auto-inflammatory episodes without normalizing the subclinical acute phase response // Eur. J. Pediatr. 2005; 164: 13–16.
2. Bodar E.J., van der Hilst J.C., Drent J.P. et al. Effect of etanercept and anakinra on inflammatory attaks in the hyper IgD syndrome: introduction a vaccination provocation model // Neth. J. Med. 2005; 63: 260–264.
3. Barron K., Athreya B., Kastner D. Periodic fever syndromes and other inherited autoinflammattory diseases. Textbook of pediatric rheumatology / Ed. by J.T. Cassidy et al. — 6th ed. — Elsevier Saunders, 2011: 642–660.
4. Cuisset L., Jeru I., Dumont B. et al and the French CAPS study group. Mutationin in the autoinflammatory cryopyrin-associated periodic syndrome gene: epidemiological study and lessons from eight years of genetic analysis in France // Ann. Rheum. Dis. 2011; 70: 495–305.
5. Dode C., Le Du N., Cuisset L. et al. New mutations of CIAS that are responsible for Muckle-Wells Syndrome and familial cold urticaria: a novel mutation underlies both syndrome // Am. J. Hum. Gen. 2002; 70: 1498–1506.
6. Drenth G., van der Meer G.W. Hereditary Periodic fever // The new England journal of medicine. 2001; 345: 1748–1757.
7. Fietta P. Autoinflammatory disease: the hereditary periodic fever syndromes // Acta Biol. Ateneo Parmense. 2004; 75: 92–99.
8. Hatemi G., Silman A., Bang D. EULAR recommendation for the management of Behcet’s disease // Ann. Rheum. Dis. 2008; 67: 1656–1662.
9. Hawkins P.L., Lachmann H.J., McDermott M.F. Interleikin-1 receptor antagonist in the Muckle-Wells syndrome // N. Engl. J. Med. 2003; 348: 2583–2584.
10. Haas D., Hoffmans G.F. Mevolonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome // Orphanet. J. of Rare Dis. 2006; 1: 13–18.
11. Hoffman H.M., Mueller J.L., Broide D.H. et al. Mutation of a new gene encoding a putative pyrin-like protein caused familial cold autoinflammatory syndrome and Muckle-Wells syndrome // Nat. Genet. 2001; 70: 301–305.
12. Hoffman H.M., Wanderer A.A., Broide D.H. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever // J. Allergy. Clin. Immunol. 2001; 108: 615–620.
13. Hoffman H.M., Throne M.L., Amar N.J. et al. Efficacy and safety of rilonacept (interleikin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies // Arthritis Rheum. 2008; 58(8): 2443–2452.
14. Hoffman H.M., Rosengren S., Boyle D.L. et al. Prevention of cold associated acute inflammation in familial cold autoinflammatory syndrome by interleikin–1 receptor antagonist // Lancet. 2004; 364: 1779–1785.
15. International Study Group for Behcet’s disease. Criteria for Diagnosis of Behcet’s disease // Lancet. 1990; 335: 1070–1080.
16. Federico G., Rigante D., Pugliese A.L. et al. Etanercept induces improvement of arthropathy in chronic infantile neurological cutaneous articular (CINCA) syndrome // Scand. J. Rheumatol. 2003; 32: 312–314.
17. Jaravello W., Pomagnoli M., Gaini R.M. Effectiveness of adenotonsillectomy of PFAPA syndrome randomized study // J. Pediatr. 2009; 155: 230–233.
18. Ruijk L.M., Hoffman H.L., Neven B., Frenkel J. Episodic autoinflammatory disorders in children // Handbook of Systemic Autoimmune Disease. — V. 6. Pediatrics in systemic autoimmune disease / Ed. by R. Cimas, T. Lehman. — Elsevier, 2008. — 119–135.
19. Kusuhara K., Nomura A., Nakao F., Hara T. Tumor necrosis factor receptor–associated periodic syndrome with a novel mutation in the TNFRSF1A gene in a Japanese family // Eur. J. Pediatr. 2004; 163: 30–32.
20. Lachmann H., Kone-Paut I., Kuemmerle-Deschner G.B. et al. For the Canakinumab in CAPS Study Group. Use of canacinumab in the cryopyrin-associated periodic syndrome // N. Engl. J. Med. 2009; 360: 2416–2425.
21. Lovell D.J., Bowyer S.L., Solinger A.M. Interleikin-1 blokade by anakinra improves clinical symptoms in patients with neonatal-onset multisystem inflammatory disease // Arthr. Rheum. 2005; 52: 1283–1286.
22. Lidar M. Livneh. Familial Mediterranean fever: clinical, molecular and management advancement // The Netherlands J. Med. 2007; 65: 318–324.
23. Ozen S., Frenkel J., Ruperto N., Gattorno M. The Eurofever Project: towards better care for autoinflammatory diseases // Eur. J. Pediatr. 2011; 170(4): 445–452.
24. Simon A., van der Meer J.W.S. Pathogenesis of familial periodic fever syndromes or hereditary inflammatory syndromes // Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007; 292: 86–98.
25. Toplak N., Frenkel J., Ozen S. et al. PRINTO, EUROFEVER and EUROTRAPS Projects. The Eurofever registry for autoinflammatory disease: results of the first 15 months of enrolment // Pediatric Rheumatology, 2011; 9(11): 115.
26. Toker O., Hashkes P.J. Critical appraisal of canakinumab in the treatment of adults and children with cryopyrin-associated periodic syndrome (CAPS) // Biologics: Targets and Therapy, 2010; 4: 131–138.
27. Yuksel S., Yalcinkaya F., Acar B. et al. Clinical improvement with infliximab in a child with amyloidosis secondary to familial Mediterranean fever // Rheumatol. 2006; 45: 1307–1308.
28. Алекберова З.С. Болезнь Бехчета. — М., 2007. — 235 с.
29. Баранова О.В., Щербина А.Ю. Аутовоспалительные заболевания: Практическое руководство по детским болезням / Под ред. В.Ф. Коколиной и А.Г. Румянцева. — Т. VIII. Иммунология детского возраста / Под ред. А.Ю. Щербиной и Е.Д. Пашановой. — М.: Медпрактика-М, 2006.
30. Ревматология: Клинические рекомендации. — 2-е изд. / Под ред. Е.Л. Насонова. — М.: ГЭЭТАР–Медиа, 2010. — 1070–1080.
31. Кузьмина Н.Н., Мовсисян Г.Р. PFAPA (periodic fever, aphthousis stomatitis, pharingitis, cervicaladinitis-периодическая лихорадка, афтозный стоматит, шейный аденит) или синдром Маршалла у детей // Науч.-практич. ревматол. 2005; 5: 80–89.
32. Кузьмина Н.Н., Федоров Е.С., Мовсисян Г.Р., Салугина С.О. Аутовоспалительные заболевания у детей — современный взгляд на проблему // Науч.-практ. ревматол. 2009; 1: 63–75.
33. Салугина С.О., Кузьмина Н.Н., Федоров Е.С. Аутовоспалительные синдромы — «новая» мультидисциплинарная проблема педиатрии и ревматологии // Педиатрия; 2012: 91, № 5; 120–132.