Статья опубликована на с. 30-32 (Укр.)
В історичному плані головну роль у формуванні бронхолегеневої дисплазії відводили високому відсотку кисню при проведенні штучної вентиляції легень (Northway W.N. Jr та співавт., 1967). При гіпероксії події розвиваються за таким сценарієм: ураження киснем → запалення → репарація за рахунок проліферації фібробластів. У подальшому звернули увагу на більш високу частоту формування БЛД при використанні значних величин «пікового тиску» під час проведення штучної вентиляції легень (Соаlson J.J. та співавт., 1995, 1998; Winter W.T. та спів–авт., 2013). Баротравма, що призводить до мікроскопічних розривів тканей дистальних відділів легень, виступає індуктором запальної відповіді. Волюмотравма припускає ураження легені об’ємом. У 1998 році доведено, що великий дихальний об’єм є основним фактором гострого ураження легень у дітей зі штучною вентиляцією легенів (Trembley L.N., Slutsky A.S., 1998; Carlo W.A., 2010). Сучасні дослідження свідчать про зв’язок механічного ураження легень із дискоординацією транскрипційних факторів, факторів росту фібробластів та ендотелію (Costa S.P., 2010). Подальше вивчення цих інтимних механізмів важливе для визначення стратегій впливу на координацію цитокінів для запобігання фіброзуванню легеневої тканини.
Сьогодні у зв’язку з виходжуванням немовлят із дуже малою та екстремально малою масою тіла в етіології БЛД переважає незрілість легеневої тканини та структур, що забезпечують дихання, запалення, пригнічення органогенезу легень.
Етіологія формування бронхо–легеневої дисплазії:
1. Нестача сурфактанту/недорозвинення легень → респіраторний дистрес-синдром → вентилятор-асоційоване ураження легень → грубий пневмофіброз (притаманний класичній формі БЛД).
2. Недоношеність (звичайно термін гестації ≤ 32 тижнів) → значне недорозвинення легень та мінімальний рівень сурфактанту в легенях → респіраторний ди–стрес-синдром → сурфактантна терапія/щадна вентиляція легень → пригнічення росту легень, легеневих судин/мінімальний пневмофіброз (більш характерні для нової форми бронхолегеневої дисплазії).
Фактори, що сприяють розвитку бронхолегеневої дисплазії:
1. Внутрішньоутробна інфекція.
2. Гіпоксія/гіперкапнія/ацидоз.
3. Генетична схильність до розвитку респіраторної патології.
4. Коморбідна патологія:
— функціонуюча артеріальна протока;
— патологія, пов’язана з порушенням нейрореспіраторного драйву.
1. Недорозвинення легень та нестача сурфактанту
Основою розвитку нової форми бронхо–легеневої дисплазії є народження дитини до появи септальних гребенів. Штучна вентиляція легень, відносно низька температура в дихальних шляхах, гіпоксія, ацидоз, у деяких випадках гіперкапнія та гіпоглікемія посилюють пригнічення правильного ходу активації ростових факторів та росту легень, легеневих судин. За відсутності сурфактанту дихальні шляхи з вузьким діаметром та альвеоли спадаються при кожному вдиху, що призводить до прогресуючого ателектазування легень. Білковий ексудат та епітеліальний дебріс (епітеліальні залишки) накопичуються в дихальних шляхах. Це веде до зменшення загального об’єму легень.
У табл. 1 подано частоту розвитку бронхолегеневої дисплазії в дітей із малою масою тіла за даними Європейського респіраторного товариства 2011, 2014 років та результати власних досліджень залежності розвитку бронхолегеневої дисплазії від маси тіла недоношеного в Харківській області (2011–2014 рр.).
2. Вентилятор-асоційоване ураження легень
2.1. Гіпероксія
Токсичні ефекти кисню були відомі з кінця XIX століття. Перші докази взаємозв’язку між токсичністю кисню й захворюваннями новонароджених було виявлено на початку 50-х років XX ст., коли описано випадки ретинопатії в недоношених дітей, які дихають киснем високої концентрації. У наступні роки відзначено значний прогрес у розумінні токсичності похідних кисню та вільних радикалів. Кисень здатний приймати електрон на свою зовнішню орбіту з утворенням вільних радикалів: супероксидного аніону (О2•–), пероксиду водню (Н2О2), ліпідного пероксиду (LOOH), гідроксильних радикалів (•ОН), а також радикалу оксиду азоту (NO•) і феноксил-радикалу (С6Н5О•).
Вільні радикали активних форм кисню можуть реагувати між собою, а утворені молекули — трансформувати інші молекули на токсичні продукти за допомогою ланцюгових реакцій. Антиоксидантний потенціал плода значно нижчий за такий у старших дітей і дорослих. Активність основних ферментів — руйнівників активних форм кисню, таких як глутатіонпероксидаза (GSH-Px), каталаза (Cat) і супероксиддисмутаза (SOD), різко знижена у глибоко недоношених дітей.
У 2000 році проведено мультицентрове плацебо-контрольоване дослідження інтратрахеального введення супероксиддисмутази недоношеним новонародженим після введення першої дози сурфактанту. Хворі на БЛД, які отримували в складі терапії супероксиддисмутазу, мали меншу потребу в респіраторній терапії порівняно з малюками, які отримували плацебо (Davis J.M., Richter S.E., 2000). Однак питання про призначення супероксиддисмутази для лікування БЛД наразі залишається дискусійним.
У недоношеного також виявляється недостатність неферментних антиоксидантних факторів. Загальна антиоксидантна активність плазми крові наростає в процесі внутрішньоматкового розвитку, але все одно виявляється низькою при народженні, особливо передчасному. Аскорбінову кислоту в періоді новонародженості не слід вважати антиоксидантним засобом. У певних умовах вона може бути прооксидантом. У періоді новонародженості, коли відзначається низький антиоксидантний потенціал, високі рівні вітаміну С можуть стати причиною несприятливого прогнозу в недоношених дітей через низьку резистентність мембран клітин життєво важливих органів.
Потужним джерелом активних форм кисню в новонародженого є нейтрофіли — безпосередні й миттєві реагенти на подразнення слизової респіраторного тракту. D.P. Carlton у дослідах на тваринах відмічає, що нейтрофіли в легенях з’являються одразу після початку штучної вентиляції легень (Carlton D.P., Albertine K.H., 2000). Активні форми кисню поряд з інфекцією, баротравмою виступають активаторами нейтрофілів, що може служити пусковим моментом запалення, деструкції та подальшої аномальної репарації легеневої тканини. Нейтрофіли та ліпідні продукти деструкції клітинних мембран активують каскад запальних реакцій із продукцією арахідонової кислоти. Вона, у свою чергу, катаболізується з утворенням цитокінів і лейкотрієнів, тромбоксану, простагландинів і простацикліну, що викликають вазодилатацію, збільшується проникність капілярів. Пропотівання альбуміну в інтерстицій змушує посилювати параметри ШВЛ, провокуючи баротравму. Активовані нейтрофіли виділяють колагеназу та еластазу, що безпосередньо руйнують тканини. Інгібітором еластази та її активації вільними радикалами кисню виступає α1-протеїназа. Її використання для запобігання розвитку БЛД у новонароджених дітей перебуває в стадії вивчення.
2.2. Механічна вентиляція
— Волюмотравма.
— Баротравма.
— Ателектатична травма.
— Травма, обумовлена гіперволемією.
2.2.1. Волюмотравма
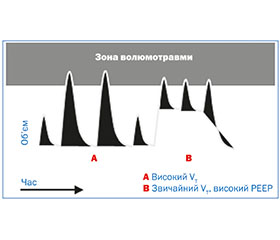
Волюмотравма виникає за рахунок перероздування легень при використанні великих дихальних об’ємів. Унаслідок волюмотравми базальна мембрана розтягується. Іноді перерозтягування веде до розриву альвеол, бронхіол та судин. Розрив альвеол і руйнування еластину, обумовлені перерозтягуванням легеневої тканини, запускають каскад запальної реакції з припливом лейкоцитів і вивільненням активних форм кисню, інтерстиціальним набряком з накопиченням води та білків.
Параметри вентиляції щодо можливого виникнення волюмотравми подано на рис. 1.
2.2.2. Баротравма
Баротравма в новонароджених зумовлена високим тиском під час штучної вентиляції легенів. Високий тиск не тільки спричинює запалення або розрив альвеол, а і гальмує альвеоляризацію й ангіо–генез. Доведено, що рестрикція грудної клітки, притаманна недоношеним, сприяє підвищенню тиску в дихальних шляхах, індукуючи баротравму (Hernandez Н. та співавт., 1988).
В експериментах учені досліджували дію на легеневу тканину високого та низького тиску, великих і малих об’ємів, щоб визначити, який із механізмів дійсно має шкідливу дію на легені недоношеного. Дане дослідження демонструвало наявність маркерів ушкодження (інтерстиціальний набряк легенів, ушкодження епітелію, формування гіалінових мембран) у групі, де застосовували високий дихальний об’єм і низький піковий тиск при штучній вентиляції легень. Так, менш травматична вентиляція з високим піковим тиском і низьким дихальним об’ємом (Hoo A., 2002).
2.2.3. Ателектатична травма
Відмінністю дихальних шляхів недоношеного вважаються знижений комплайєнс між потребою у вентиляції та об’ємом дихальних шляхів, дефіцит сурфактанту, обумовленого недоліком синтезу або виснаженням його запасів. Розправившись після першого вдиху, альвеоли недоношеної дитини знову злипаються. Цикли альвеолярного колапсу повторюються та значно травмують паренхіму легенів недоношеного новонародженого, що призводить до тяжких респіраторних порушень. Тому однією з вимог до респіраторної підтримки недоношених є збереження оптимальної залишкової ємності легень.
2.2.4. Травма, обумовлена гіперволемією
Умови вентиляції легень недоношених із респіраторними розладами сприяють постійному підвищенню внутрішньолегеневого тиску та накопиченню рідини, що накладається на анатомічні особливості. Підвищений внутрішньолегеневий тиск стискає праву лімфатичну протоку, уповільнюючи відтік лімфи з легень. При високому альвеолярному тиску стискаються і легеневі капіляри. Це значно підсилює фільтрацію води з артеріальної частини капіляра в інтерстицій, особливо з екстраальвеолярних судин, де тиск вищий за легеневі капіляри. Указані процеси призводять до периваскулярного накопичення рідини у вигляді муфт навколо капілярів. Гіперволемія підвищує ризик ектравазації, особливо при збільшенні рСО2.
Тенденції сучасної респіраторної терапії направлені на мінімізацію гіпероксії та механічного ураження легень. Нарівні з цим експозиція кисню та механічна травма продовжує відігравати роль у пато–генезі нової БЛД.
3. Внутрішньоутробна та неонатальна інфекція
Половина всіх народжень недоношених дітей відбувається внаслідок розриву амніотичних оболонок та початку передчасних пологів (Goldenberg R.L., 2000). 20 % передчасних пологів обумовлені інфекцією в матері або патологією ембріона. У 73 % матерів зі звичними передчасними пологами до 30-го тижня гестації та у 83 % породіль, які вже мали новонароджених із масою тіла при народженні ≤ 1000 г, було виділено позитивні культури з хоріоамніону (Hartling L., 2012).
Місцева запальна реакція матки супроводжується синтезом прозапальних цитокінів (TNF-α, інтерлейкіни (IL) 1, 6, 8, 11). Цитокіни індукують простагландини Е2, викликаючи скорочення мускулатури матки та передчасні пологи. Біологічно активні речовини стимулюють цервікальне дозрівання та передчасні пологи з розривом плідних оболонок (Кетлинский С.А., 2008). IL-1 та TNF-α через активацію матричної металопротеїнази стимулюють тканинне ремоделювання та неоангіогенез (Lecart C. та співавт., 2000). Запалення гальмує нормальний морфогенез легенів. Розчинні медіатори запалення, присутні в легенях пацієнтів, порушують експресію декількох генів, критичних для розвитку. Інгібування FGF-10-запальної сигналізації включає в себе –NF-kB-залежні взає–модії між SP3- і FGF-10-промоутерами. Тому NF-kB-активація при БЛД призводить до залучення інгібуючих факторів для конкретних генних стимуляторів запалення (Carver B.J., Plosa E.J., Stinnett A.M., 2013).
Ті ж механізми обумовлюють затримку внутрішньоутробного розвитку плода з негативним впливом на фізіологічну послідовність факторів пульмонального росту та диференціювання, порушенням альвеоляризації та васкуляризації. У роботах учених США в 2010 році доведено роль запалення у пригніченні альвеоляризації в легенях пацієнтів із БЛД (Wigglesworth J.S., 2010).
Вивчається роль Ureaplasma urealiticum у розвитку бронхолегеневої дисплазії. Деякі автори вважають, що уреаплазменна інфекція не асоціюється з БЛД (Heggie А. та співавт., 2001). Інші свідчать, що тривала персистенція сприяє формуванню бронхолегеневої дисплазії (Castro-Alcaraz Р. та співавт., 2002). Виявлено, що неонатальний сепсис асоціюється з БЛД (Van Marter К. та співавт., 2002). Продовжуються дослідження щодо впливу респіраторної інфекції, а саме викликаної респіраторно-синцитіальним вірусом, аденовірусом, цитомегаловірусом, на розвиток БЛД.
За даними Харківського центру діагностики та лікування БЛД, частота неонатальної пневмонії серед дітей із БЛД становила 80,28 ± 4,75 %. Неонатальний сепсис у структурі дітей із бронхолегеневою дисплазією становив 28,6 ± 1,9 %.
4. Гіпоксія, та/або гіперкапнія, та/або ацидоз
Фактором розвитку бронхолегеневої дисплазії, що веде до гальмування онтогенезу легень, є гіпоксія. Фізіологічна гіпоксія у внутрішньоутробному періоді стимулює ростові сигнали при онто–генезі. При зниженні рО2 менше граничних показників, що спостерігається при хронічній гіпоксії плода, виникають діаметрально протилежні зрушення балансу цитокінів, гіпоксіяіндукованого фактора 1 (HIF-1) та ростових факторів (VEGF, FGF і TGF-β1). Тривала патологічна гіпоксія сприяє активації анаеробного гліколізу, централізації кровообігу, наростанню ацидозу. Це призводить до затримки внутрішньоутробного розвит–ку плода, порушення розвитку легень і гальмування процесів альвеоляризації. У позаутробному періоді гіпоксія та ацидоз гальмує синтез сурфактанту, сприяє підвищенню тиску в легеневій артерії, затримує рідину в легеневому інтерстиції, що подовжує киснезалежність ново–народженого.
5. Генетична схильність до розвитку респіраторної патології
Детермінізм бронхолегеневих захворювань досі залишається відкритим. Проте невирішене питання високого рівня клінічної варіативності БЛД серед індивідуумів у популяції, які мають однаковий гестаційний вік і помірний ятрогенний вплив під час реанімації. Для пояснення даної гетерогенності розглядаються варіанти менделевського та полігенного патерна наслідування захворювання.
На сьогодні накопичено достатньо епідеміологічних доказів того, що генетичні фактори відіграють важливу роль у розвитку багатьох хронічних захворювань бронхів та легень. Доведено, що коефіцієнт відповідності по респіраторній патології вищий у монозиготних близнюків, ніж у дизиготних, що передбачає органічний зв’язок із генетичною інформацією організму щодо сприятливості до даного захворювання. Вченими Фінляндії проведений аналіз ДНК 266 пар одностатевих близнюків, серед яких хоча б в одного із пари мав місце респіраторний дистрес-синдром новонароджених. Виявлена вірогідно частіша наявність Ile131Thr алелі у дітей, які мали РДС новонародженого (р = 0,003). У чотирьох парах тільки в одного з близнюків виявлена алель Ile131Thr. Саме ці діти з пари мали респіраторні розлади в неонатальному періоді (Ramet M., Haataja R., 2000). Відомі генетичні дефекти синтезу протеїнів і ліпідів сурфактанту пов’язані з формуванням бронхолегеневої дисплазії. На моделях тварин було показано, що експресія факторів росту фібробласту, Bmp-4 та Nkx2.1 обов’язкова для раннього розвитку легень (Perl A.K., Whittsett J.A., 2000). Описано, що дистальні відділи легень схильні до дуже рідкісних рецесивних делеційних мутацій гена Sp-B та домінантних мутацій, пов’язаних із аберантними протеїнами Sp-B (Sun H. та співавт., 2013). Пульмональна гіпо–плазія асоційована з геном DHCR7, пульмональна секвестрація — з Fox-b5 (Dani S., 2008). Вченими Японського педіатричного суспільства у 2012 році виявлений зв’язок поліморфізму кодону-54 манозозв’язуючого лектину, антагоніста рецептора гена інтерлейкіну-1 з частотою формування бронхолегеневої дисплазії (Cakmak B.C., Calkavur S., Ozkinay F. та співавт., 2012). В Україні вивчається роль генетичного поліморфізму секретоглобіну SCGB1A1 у розвитку бронхіальної астми та рецидивуючого бронхіту в дітей (Малая Н.К., Каладзе Н.Н., 2014).
У фізіологічному сенсі на етапі розвит–ку та при сформованій БЛД особливість ремоделювання легень і судин є атрибутом клітинних та позаклітинних регуляторних процесів, що забезпечуються молекулярними індукторами, регуляторами та сигнальними системами, що детермінують процеси кардіореспіраторного ремоделювання. Клітинна та/або внутрішньоклітинна експресія, а також рівень презентації специфічних рецепторів визначається тонкими генетичними механізмами. Подальше вивчення цих інтимних механізмів важливе для запобігання фіброзуванню легеневої тканини.
У зв’язку з цим протягом декількох десятиліть продовжує підвищуватися інтерес до матриксних металопротеїназ (ММП) — клітинних ензимів, що залучають позаклітинний матрикс у процеси структурно-функціонального ремоделювання легень шляхом деградації ланцюгів колагену. Останніми роками значну увагу приділяють ММП-1 як сироватковому маркеру фіброзу легень за рахунок тісної кореляції між металопротеїназами, їх ендогенними інгібіторами (Давидова И.Ф., 2010). Проведені дослідження системи «протеоліз — антипротеоліз» із визначенням вмісту серинових і цистеїнових протеїназ та їх інгібіторів, що свідчать про важливість оцінки їх балансу в системі протеолізу при БЛД (Черненко Л.М., 2012). Тому протеоліз сьогодні розглядається як особлива форма біологічного контролю при бронхолегеневій дисплазії, займає центральне місце в реалізації різноманітних біохімічних процесів та швидкої фізіологічної відповіді на умови, що змінюються. ММП-1 здатна специфічно гідролізувати основні компоненти екстрацелюлярного матриксу, інтерстиційний колаген I, II, III типів у спіральній ділянці на особливих потрійних ділянках у 3/4 частини N-кінцевого домену, відтворюючи 3/4 та 1/4 фрагменту колагену, що є стійкими до дії більшості протеїназ. ММП-1 гідролізує мінорні колагени VII та X типів, желатини колагенів, лінк-протеїн хряща, а також білки сполучнотканинного матриксу: ектактин, агрикан, казеїн, альфа-2-макроглобулін, синтетичні субстрати, які за своєю амінокислотною послідовністю відповідають ділянці в структурі колагену та альфа-2-
макроглобуліну, що гідролізується. ММП-1 спричиняє руйнування еластичного каркасу тканин та порушення нормальної архітектоніки легень. Тонкі еластичні волокна міжальвеолярних перетинок руйнуються швидше, ніж пучки в стінках, — формується емфізема легень. Виникає обструктивний синдром, в основі якого лежить порушення еластичної напруги між легеневою паренхімою і бронхами. Синтез та секреція ММП-1 здійснюється під дією прозапальних цитокінів, інтегринів, форболових ефірів, ліпополісахаридів, колхіцину, простагландину Е (PGE), а головним джерелом вважаються активовані макрофаги, нейтрофіли, фібробласти. Активність матриксної метало–протеїнази-1 регулюється на різних рівнях, включаючи транскрипцію, активацію білка та взаємо–дію з ендогенними інгібіторами в легеневому інтерстиції.
Унікальною особливістю ММП-1 є здатність ініціювати та продовжувати «ураження» проміжного колагену. Проте активність ММП-1 визначається не стільки рівнем ферменту крові, скільки здатністю гена до її експресії. Поліморфізм гена матриксної металопротеїнази-1 широко вивчається в онкології та корелює з інвазією пухлини в судини, метастазами в лімфатичних вузлах та несприятливим прогнозом (Базьяк Я.Н., Жибловська К.И., 2002). Описана наявність гіперсекреторних мутацій гена ММП-1 у хворих із різними формами професійної бронхолегеневої патології (професійний хронічний бронхіт, пневмоконіози, професійна бронхіальна астма) (Фомина В.С., 2010). Водночас робіт щодо ролі експресії гена матриксної металопротеїнази-1 при бронхолегеневій дисплазії нами не знай–дено.
На кафедрі педіатрії № 1 та неонато–логії Харківського національного медичного університету (зав. кафедри — Г.С. Сенаторова) в Обласному центрі діагностики та лікування бронхолегеневої дисплазії у дітей Харківської обласної дитячої лікарні (головний лікар — Г.Р. Муратов) проведено дослідження з удосконалення діагностики формування бронхолегеневої дисплазії шляхом визначення ролі генетичних та середовищних факторів у формуванні бронхолегеневої дисплазії у новонароджених. Під спостереженням знаходились 60 близнюків/30 пар (24,1 ± 2,7 %): 54 пацієнти (90,0 ± 3,9 %) з діагнозом «бронхолегенева дисплазія» (основна група) та 6 спостережених (90,0 ± 3,9 %), які були народжені недоношеними, мали респіраторні розлади, але без бронхолегеневої дисплазії (група порівняння). Всі 6 близнюків групи порівняння були з пар, де одна дитина страждала від БЛД. Для оцінки відносної ролі спадкових та середовищних факторів використовували близнюковий метод. Визначали парну конкордантність, обчислювалась частка спадковості у формуванні БЛД за формулою Хольцингера, оцінювалась роль середовищних факторів. Поліморфізм гена матриксної металопротеїнази-1 визначався полімеразною ланцюговою реакцією (ПЛР) діагностичними наборами «SNP-експрес» виробництва НПФ «Літех», методом алельної дискримінації. За допомогою реагенту «ДНК-Експрес» відділялась ДНК із букального епітелію. Обраховувались частоти алелей та частоти алельних сполучень та їх відповідність рівновазі Харді — Вайнберга за критерієм χ2. Детекція поліморфізму проводилась методом горизонтального електрофорезу.
Виявлена різниця в групах тільки серед монозиготних близнюків жіночої статі (р < 0,05), що, з нашої точки зору, потребує подальших досліджень.
Парна конкордантність за БЛД становила 0,8, що свідчило про вагомість спадкового впливу на розвиток захворювання. 24 пари (80,0 ± 7,4 %) були конкордантні за БЛД, 6 пар (20,0 ± 7,4 %) — дискордантні. Конкордантних пар із формування бронхолегеневої дисплазії було вірогідно більше (р < 0,0001), що свідчило про можливий вплив спадкових факторів на формування захворювання. Виявлено, що із 16 пар монозиготних близнюків 9 пар були конкордантні за БЛД у неонатальному періоді (парна конкордантність — 0,562). Із 10 пар дизиготних близнюків 7 пар — конкордантні за БЛД (парна конкордантність — 0,7). Частка спадковості (0,46) за формулою Хольцингера знаходилась в інтервалі від 0,3 до 0,7, а частка впливу факторів середовища становила 0,54, що свідчило про рівну роль спадкових і середовищних факторів у формуванні БЛД.
Під час дослідження доведений рівноцінний вплив середовищних та генетичних факторів на формування БЛД, що обумовлювало необхідність вивчення алельних варіантів поліморфізму генів щодо можливості прогнозування цього хронічного захворювання під час вагітності та в новонароджених. 60 близнюків були обстежені на наявність тригерного поліморфізму (інерції) гена матриксної металопротеїнази-1. Поліморфізм гена ММП-1 (1607insG) впливав на схильність індивідуума до бронхолегеневої дисплазії (KW = H (n = 58) = 18,85; р = 0,0001). Для дітей із бронхолегеневою дисплазією було характерне переважання домінантних гомозигот (АА) та гетерозигот (Аа) за інсерцісю гуаніну у 1607 положенні (р < 0,001), що, ймовірно, були підґрунтям до підвищеної експресії ММП-1, притаманної дітям із БЛД.
Таким чином, у дітей із БЛД визначена висока ймовірність неменделевського, полігенного наслідування схильності до бронхолегеневої дисплазії, що підтверджується помірним порушенням рівняння Харді — Вайнберга.
6. Інші фактори,що можуть впливати на формування БЛД
Надмірно податлива грудна клітка. Більша величина від’ємного тиску, що генерується для розправлення дихальних шляхів, що спалися, викликає появу втягувань та деформації грудної клітки з незрілими структурами, які формують її каркас (замість розправлення ригідних легень).
Порушення механіки дихання. Внутрішньоутробні дихальні рухи мають надзвичайне фізіологічне значення, вони сприяють притоку крові до серця плода, стимуляції альвеоляризації легеневої тканини й росту судин. Деформація каркасу грудної клітки, обумовлена природженими вадами розвитку скелета, діафрагмальною грижею, кістами, пухлинами грудної порожнини, обмежує внутрішньо–утробні дихальні рухи та ріст легень.
Питання порушення центральної регуляції дихання обумовлені частим сполученням їх із бронхолегеневою дисплазією в пацієнтів, які знаходяться під спостереженням у центрах перинатальної патології. Пацієнти з ураженням ЦНС мають рецидивуючі респіраторні проблеми. Експерименти новозеландських учених у 2007 році довели, що ушкодження провідних шляхів на рівні шийного відділу спинного мозку в третьому триместрі вагітності веде до зниження маси легенів при народженні та зменшення сумарної дезоксирибонуклеїнової кислоти (ДНК) у дистальному відділі респіраторного тракту. Вчені дійшли висновку, що патогенетичні основи дисплазії легень у таких дітей полягають у відсутності адекватної тяги, що гальмує альвеоляризацію респіраторного тракту. Доведені єдині генетичні основи порушення експресії гена VEGF на ендотелії легеневих судин і судин центральної нервової системи у плода людини, що може об’єднувати в патологічні механізми респіраторну та нервову систему. Наочним прикладом є brain-lung-thyroid syndrome, обумовлений мутацією гена Nkx2.1 та тиреоїдного трансформуючого фактора з клінічними проявами гіпотиреозу, респіраторного дистрес-синдрому та хореї.

Пройти тестовое задание к симпозиуму

/30_u.jpg)
/31_u.jpg)