Журнал «Здоровье ребенка» 7 (67) 2015
Синдром Ангельмана. Часть 3 (дифференциальная диагностика и лечение)
Резюме
Статья посвящена проблеме дифференциальной диагностики и лечения больных с синдромом Ангельмана (AS). Представлены наиболее часто встречающиеся синдромы, имеющие сходные с AS фенотипические проявления, приведены основные дифференциальные отличия AS-подобных синдромов с указанием причинно-значимого гена. Большое внимание уделено основным направлениям лечения больных с синдромом Ангельмана: организации режима, посиндромной терапии, оказанию психологической, логопедической, ортопедической помощи, трудотерапии, мероприятиям, направленным на социальную адаптацию. Статья содержит новейшую информацию о современных прогрессивных направлениях медикаментозного управления AS. Для оптимизации диагностического и терапевтического процессов приведены данные о международных фондах синдрома Ангельмана.
Стаття присвячена проблемі диференційної діагностики та лікування хворих iз синдромом Ангельмана (AS). Представлені синдроми, що зустрiчаються найчастіше, які мають схожі з AS фенотипічні прояви, наведені основні диференційні відмінності AS-подібних синдромів із зазначенням причинно-значущого гена. Велику увагу приділено основним напрямкам лікування хворих iз синдромом Ангельмана: організації режиму, посиндромнiй терапії, наданню психологічної, логопедичної, ортопедичної допомоги, трудотерапії, заходам, спрямованим на соціальну адаптацію. Стаття містить новітню інформацію про сучасні прогресивні напрямки медикаментозного управління AS. Для оптимізації діагностичного та терапевтичного процесів наведені дані про міжнародні фонди синдрому Ангельмана.
The article discusses the problem of differential diagnosis and treatment of patients with Angelman syndrome (AS). It provides the most common syndromes with similar to AS phenotypes, the main differences between AS-like syndromes, indicating the causative gene. Much attention is given to the basic directions of treating patients with Angelman syndrome: organization of regime, syndromic treatment, providing psychological, speech therapy, orthopedic services, occupational therapy, activities aimed at social adaptation. This article contains the latest information about modern progressive directions of AS medical management. To optimize the diagnostic and therapeutic process, data about international Angelman syndrome foundations are shown.
Ключевые слова
синдром Ангельмана.
синдром Ангельмана.
Angelman syndrome.
Статья опубликована на с. 81-87
Дифференциальная диагностика синдрома Ангельмана
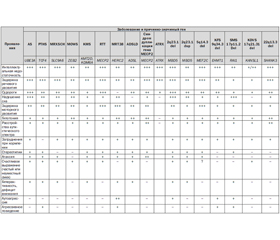
Дифференциальная диагностика синдрома Ангельмана (AS) проводится с различными синдромами, которые имеют фенотипически похожие проявления (табл. 1) [32, 43].
Синдром Питта — Хопкинса (Pitt-Hopkins syndrome, PTHS; OMIM #610954). У больных с данным синдромом отмечаются задержка развития речи с умственной отсталостью, микроцефалия, судороги, широкий рот, веселый нрав. Отличительной особенностью является возникновение эпизодических пароксизмов гипервентиляции и/или задержки дыхания во время бодрствования (у 55–60 %). В основе лежат мутации гена транскрипционного фактора 4 TCF4 [4].
Проявления синдрома Кристиансона (Christianson syndrome, MRXSCH; OMIM #300243), или синдрома X-сцепленной ментальной ретардации, могут напоминать фенотип AS. Так, для больных с синдромом Кристиансона характерны умственная отсталость, немота, постнатальная микроцефалия, атаксия, гиперкинезы, судорожный синдром, веселый нрав. В основе заболевания лежат мутации гена катион/протонного антипортера 6 SLC9A6 (solute carrier family 9, subfamily A (NHE6, cation proton antiporter 6), member 6). Тип наследования — X-сцепленный [34].
У больных с синдромом Моуата — Вильсон (Mowat-Wilson syndrome, MOWS; OMIM #235730), как и у лиц с AS, наблюдаются умственная отсталость, микроцефалия, эпилепсия, открытый рот, выступающий заостренный подбородок, счастливое выражение лица. Отличительными признаками являются гипертелоризм глазных яблок, сходяще–еся косоглазие, закругленный кончик носа и повернутая назад ушная раковина. Развитие заболевания обусловлено мутациями гена ZEB2 (zinc finger E-box binding homeobox 2) [2, 13].
У больных с синдромом Кабуки (Kabuki syndrome 1; Kabuki make-up syndrome; KMS; OMIM #147920), или синдромом Ниикава — Куроки (Niikawa-Kuroki syndrome), наблюдаются признаки, близкие к проявлениям AS: легкий или умеренный интеллектуальный дефицит, задержка речевого развития, мышечная гипотония, нарушения аутистического спектра, эпилепсия, нарушения координации, моторная неловкость. Отличительным фенотипическим признаком KMS является выворот латеральной части нижнего века, что вместе с аркообразными бровями придает пациентам схожесть с актерами японского театра Кабуки. У больных также отмечаются микроцефалия, гипертелоризм глазных яблок, длинные глазные щели, длинные и густые ресницы, сплющенный и широкий кончик носа с вывернутыми вперед ноздрями, готическое небо, гиподонтия, широкие межзубные промежутки. Синдром ассоциирован с мутациями гена гистоновой метилтрансферазы KMT2D (lysine (K)-specific methyltransferase 2D) и гена гистоновой деметилазы KDM6A (lysine (K)-specific demethylase 6A) [5, 16].
Клинические проявления у лица женского пола с AS могут напоминать синдром Ретта (Rett syndrome, RTT; OMIM #312750), для которого характерны приобретенная микроцефалия, тяжелая задержка речевого развития, судороги. Однако при синдроме Ретта у больных нет проявлений «счастливости». Регрессирование умственного развития у них манифестирует после 6–18 месяцев жизни и сопровождается приступами крика, плача, безутешности, паники, аутистическими расстройствами. Эпизодически возникает апноэ и прогрессирует апраксия. Заболевание связано с мутациями гена метилсвязывающего протеина 2 MECP2 (methyl CpG binding protein 2). Тип наследования — X-сцепленный [12].
Синдром аутосомно-рецессивной ментальной ретардации 38 (mental retardation, autosomal recessive 38, MRT38; OMIM #615516), или синдром дефицита HERC2, проявляется задержкой умственного, психического, речевого, моторного развития, а также социальных функций. Для данной группы больных характерны: мышечная гипотония, затруднения вскармливания на первом году жизни, нарушения аутистического спектра, приступы агрессии, членовредительства, импульсивность. Отличительным признаком является голубой цвет радужной оболочки. Заболевание ассоциировано с гомозиготной миссенс-мутацией гена HERC2 (HECT and RLD domain containing E3 ubiquitin protein ligase 2) [38, 43].
Дефицит аденилсукциназы (adenylosuccinase deficiency, ADSLD; OMIM #103050) по клиническим проявлениям очень близок к фенотипу AS. Выражены нарушения аутистического спектра, включая избегание контакта взглядом «глаза в глаза». Отличительными особенностями являются умеренная микроцефалия, брахицефалия, плоский затылок, метопический шов, расходящееся косоглазие, маленький нос с вывернутыми ноздрями, тонкая верхняя губа, широкий рот и низко посаженные ушные раковины. Диагноз верифицируют на основании определения сукцинил-аминоимидазол-карбоксамида рибозида (SAICAr) и сукциниладенозина (S-Ado) во внеклеточных жидкостях (сыворотка крови, спинномозговая жидкость или моча). В основе заболевания лежат мутации гена аденилсукциназы (EC 4.3.2.2) ADSL (adenylosuccinate lyase) [23].
Синдром дупликации гена MECP2 (MECP2 duplication syndrome) наблюдается только у лиц мужского пола, у которых отмечаются мышечная гипотония с раннего возраста, тяжелая умственная отсталость, выраженное отставание речевого развития вплоть до отсутствия речи, нарушения аутистического спектра, эпилепсия. В отличие от AS для синдрома дупликации гена MECP2 характерны прогрессивная спастическая миопатия, рецидивирующие инфекции. Заболевание связано с дупликации гена MECP2. Большинство случаев наследуются по Х-сцепленному типу, однако были документированы и de novo мутации (спорадические) случаи [36, 39].
Синдром аутосомно-доминантной ментальной ретардации 1 (mental retardation, autosomal dominant 1 syndrome; MRD1; OMIM #156200), синдром псевдо-Ангельмана, или синдром делеции хромосомы 2q23.1, и синдром дупликации хромосомы 2q23.1 могут проявляться AS-подобным фенотипом. Но у данных больных идентифицируется нарушение локуса хромосомы 2q23.1, в котором расположен ген метилсвязывающего протеина 5 MBD5 (methyl-CpG binding domain protein 5) [28–30].
Синдром Клифстра (Kleefstra syndrome, KFS; OMIM #610253), или синдром делеции хромосомы 9q34.3, имеет клинические признаки, схожие с таковыми при AS. У больных отмечаются умеренная или тяжелая интеллектуальная недостаточность с минимальным развитием речи, мышечная гипотония, особенно в раннем детском возрасте, нарушения сна с частыми пробуждениями ночью или под утро, нарушения аутистического спектра, гиперактивность с дефицитом внимания, моторная неловкость, атаксия, приступы судорог. Характерными проявлениями KFS считают: гипоплазию средней части лица, плоское лицо, гипертелоризм глазных яблок, синофриз, короткий вздернутый нос, прогнатизм, вывернутую нижнюю мясистую губу, приоткрытый рот, приступы аутоагрессии и агрессивного поведения. В основе заболевания лежит делеция региона q34.3 хромосомы 9, где расположен ген гистоновой метилтрансферазы EHMT1 (euchromatic histone-lysine N-methyltransferase 1) [24].
Дети с синдромом Смит — Магенис (Smith-Magenis syndrome, SMS; OMIM #182290), или синдромом делеции хромосомы 17p11.2, уже на первом году отстают в физическом и психомоторном развитии. В последующем у них наблюдается интеллектуальная недостаточность, задержка речевого развития (больше страдает моторная, чем сенсорная речь), судороги, нарушение сна (очень раннее пробуждение и выраженная сонливость в течение дня), расстройства аутистического спектра. Специ–фическими признаками фенотипа являются плоское широкое лицо, гипоплазия средней части лица, выпуклый лоб, приоткрытый рот. Для пациентов характерна инверсия суточного ритма секреции мелатонина, аутоагрессия. В основе заболевания лежит делеция региона p11.2 хромосомы 17, где расположен ген протеина 1, индуцированного ретиноевой кислотой, RAI1 (retinoic acid induced 1) [1, 3, 15].
Синдром Кулена — де Фриза (Koolen-De Vries syndrome, KDVS; OMIM #610443), или синдром делеции хромосомы 17q21.31, гаплонедостаточности гена KANSL1, проявляется мышечной гипотонией, умственной отсталостью от легкой до умеренной степени тяжести, задержкой речевого развития, пароксизмами судорог и дружелюбием. У больных с синдромом KDVS наблюдаются: удлиненное лицо, высокий лоб, большие оттопыренные ушные раковины, косой разрез глазных щелей, эпикант, грушевидный нос, врожденные пороки сердца (стеноз легочной артерии, дефекты перегородок и двустворчатый аортальный клапан), гипермобильность суставов, аномалии волос, кожи и зубов, крипторхизм, гипоспадия, пузырно-мочеточниковый рефлюкс. В основе заболевания лежат делеции региона q21.31 хромосомы 17, в котором располагается ген KANSL1 (KAT8 regulatory NSL complex subunit 1) [17, 26].
Синдром Фелан — Мак-Дермид (Phelan-McDermid syndrome; OMIM #606232), или синдром делеции хромосомы 22q13.3, также характеризуется наличием у больных умственной недостаточности, задержки развития вплоть до отсутствия речи, расстройств аутистического спектра, но в отличие от синдрома Ангельмана наблюдается выраженная неонатальная гипотония, большие мясистые руки, диспластические ногти и снижение активности потоотделения. Причиной заболевания является делеция 22q13.3 или мутация гена SHANK3 (SH3 and multiple ankyrin repeat domains 3) [25, 37].
При проведении дифференциальной диагностики также должны быть учтены AS-подобные синдромы: синдром гаплонедостаточности гена FOXG1 (FOXG1 haploinsufficiency syndrome) [7], синдром гаплонедостаточности гена STXBP1 (STXBP1 haploinsufficiency syndrome) [11], синдром гаплонедостаточности гена MEF2C (MEF2C haploinsufficiency syndrome) [31] и синдром X-сцепленной альфа-талассемии и интеллектуальной недостаточности (alpha-thalassemia/intellectual disability syndrome, ATRX; OMIM #301040) [41].
Терапия
Основными направлениями лечения являются: организация режима, посиндромная терапия, оказание психологической, логопедической, ортопедической помощи, трудотерапия, мероприятия, направленные на социальную адаптацию [20, 21].
Организация режима и психологическая поддержка
В периоде грудного возраста особое внимание уделяется индивидуализации режимов вскармливания грудью. При неэффективности изменения техник грудного вскармливания рекомендуется вскармливание с использованием соски. Могут потребоваться специальные соски и другие стратегии управления слабого или несогласованного сосания [45]. У детей старшего возраста необходимо организовывать жизненное пространство с учетом их моторных особенностей (безопасная мебель, эргометрическое расположение мебели, индивидуальные по форме и особо устойчивые стулья и т.д.). Особенностью логопедической помощи больным с AS считают ее направленность на невербальные методы общения [45].
Медикаментозная терапия
Основными целями медикаментозного лечения являются контроль эпилептических приступов, коррекция расстройств сна и поведенческих нарушений.
Этиологическая терапия
Этиологического лечения AS не существует. В настоящее время разрабатываются новые лекарственные средства, которые способствовали бы усилению экспрессии гена UBE3A. В частности, синтезированы антисмысловые олигонуклеотиды, обусловливающие снижение уровня содержания транскрипта UBE3A-АТС. В экспериментальных условиях показано, что при их применении уменьшается некоторый когнитивный дефицит [27]. Изучаются ингибиторы топоизомеразы II (топотекан, иринотекан, этопозид и дексразоксан/ICRF-187), снимающие сайленсинг гена UBE3A на отцовской хромосоме. Продемонстрировано, что топотекан в наномолярной концентрации повышает каталитическую активность протеина UBE3A в нейронах мышей с UBE3A-нулевой копией материнской хромосомы. Топотекан также подавляет экспрессию антисмыслового транскрипта UBE3A-ATC. Повышенная экспрессия гена UBE3A в нейронах спинного мозга мышей наблюдается на протяжении не менее 12 недель после прекращения лечения топотеканом [6, 22].
Попытка использования диетических добавок (бетаин, метафолин, креатин и витамин B12) с целью подавления продукции антисмыслового транскрипта UBE3A-АТС и усиления экспрессии гена UBE3A не привела к изменению сайт-специфического метилирования ДНК и не способствовала уменьшению тяжести проявления AS [9]. Хотя у некоторых пациентов при назначении бетаина и фолиевой кислоты наблюдалась тенденция к улучшению состояния [35].
Одним из прогрессивных направлений медикаментозного управления AS является терапия, способствующая восстановлению дендритных отростков нейронов. Так, было установлено, что миноциклина гидрохлорид, представляющий группу тетрациклиновых антибиотиков, который легко проникает через гематоэнцефалический барьер, восстанавливает синаптические функции через модуляцию структуры дендритных шипов. Миноциклин изменяет морфологию дендритных шипов нейронов гиппокампа, обусловливая формирование незрелых удлиненных в зрелые грибовидные шипы, что сопровождается восстановлением синаптической пластичности и улучшением пространственной памяти [8, 18]. Назначение миноциклина детям с AS вызывает достоверное улучшение адаптивного поведения [19].
Противосудорожная терапия
Несмотря на многочисленные исследовательские работы, посвященные разработке стратегии медикаментозного контроля за развитием эпиприступов у больных с AS, не существует единого критерия, который позволял бы выбрать оптимальный противоэпилептический препарат. При лечении судорожного синдрома у больных с AS широко используют препараты вальпроевой кислоты, топирамата, ламотриджина, леветирацетама, клоназепама, менее часто назначают препараты карбамазепина, этосуксимида, фенитоина, фенобарбитала [33, 44]. Фенитоин, карбамазепин, окскарбазепин и вигабатрин в монотерапии могут приводить к аггравации эпилептических приступов [1].
Лечение нарушений сна
Для лечения нарушений сна рекомендуют применение дифенилгидрамина [42], введение мелатонина (0,3 мг) за один час до сна [10, 40]. Использование седативных средств (фенотиазины и атипичные антипсихотики) не показано, так как они вызывают негативные эффекты [14].
Терапия при слюнотечении
Лечение слюнотечения — оперативное (реимплантация слюнных протоков) или местное (применение лекарственных средств, подавляющих слюнообразование, например скополамина) — не эффективно.
Медицинские фонды синдрома Ангельмана
При организации диагностического и терапевтического процессов рекомендуется воспользоваться помощью фондов синдрома Ангельмана [14]:
Angelman Syndrome Foundation, Inc. (ASF)
4255 Westbrook Drive
Suite 219
Aurora IL 60504
Телефон: 800-432-6435 (toll-free); 630-978-4245
Факс: 630-978-7408
Электронная почта: info@angelman.org
Веб-сайт: www.angelman.org
Foundation for Angelman Syndrome Therapeutics (FAST)
PO Box 608
Downers Grove IL 60515
Телефон: 866-783-0078; 630-852-3278
Факс: 630-852-3270
Веб-сайт: http://www.cureangelman.org/default.html
Список литературы
1. Новоселова О.Г. Перспективы диагностики расстройств аутистического спектра у детей / О.Г. Новоселова, Г.А. Каркашадзе, Н.В. Журкова, О.И. Маслова // Вопросы современной педиатрии. 2014; 13 (3): 61-68.
2. Adam M.P., Conta J., Bean L.J.H. Mowat-Wilson Syndrome // Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors // GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2007 Mar 28 [updated 2013 Nov 26]. PMID: 20301585.
3. Angriman M. Sleep in children with neurodevelopmental di–sabilities / M. Angriman, B. Caravale, L. Novelli, R. Ferri, O. Bruni // Neuropediatrics. 2015 Jun; 46(3): 199-210. doi: 10.1055/s-0035-1550151.
4. Ardinger H.H., Welsh H.I., Saunders C.J. Pitt-Hopkins Syndrome // Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors // GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2012 Aug 30.
5. Arnaud M. Le syndrome Kabuki: mise au point et revue de la litterature / M. Arnaud, M. Barat-Houari, V. Gatinois et al. // Archives de Pediatrie. 2015 Jun; 22(6): 653-60. doi: 10.1016/j.arcped.2015.03.020.
6. Beaudet A.L. Angelman syndrome: Drugs to awaken a paternal gene // Nature. 2011 Dec 21; 481(7380): 150-2. doi: 10.1038/nature10784.
7. Bertossi C. Forkhead box G1 gene haploinsufficiency: an emer–ging cause of dyskinetic encephalopathy of infancy / C. Bertossi, M. Cassina, A. Cappellari et al. // Neuropediatrics. 2015 Feb; 46(1): 56-64. doi: 10.1055/s-0034-1395345.
8. Bilousova T.V. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model / T.V. Bilousova, L. Dansie, M. Ngo, J. Aye, J.R. Charles, D.W. Ethell, I.M. Ethell // J. Med. Genet. 2009 Feb; 46(2): 94-102. doi: 10.1136/jmg.2008.061796.
9. Bird L.M. A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome / L.M. Bird, W.H. Tan, C.A. Bacino et al. // Am. J. Med. Genet. A. 2011 Dec; 155A(12): 2956-63. doi: 10.1002/ajmg.a.34297.
10. Braam W. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial / W. Braam, R. Didden, M.G. Smits, L.M. Curfs // J. Child Neurol. 2008 Jun; 23(6): 649-54. doi: 10.1177/0883073808314153.
11. Campbell I.M. Novel 9q34.11 gene deletions encompassing combinations of four Mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A / I.M. Campbell, S.A. Yatsenko, P. Hixson et al. // Genet. Med. 2012 Oct; 14(10): 868-76. doi: 10.1038/gim.2 012.65.
12. Christodoulou J., Ho G. MECP2-Related Disorders // Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors // GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2001 Oct 03 [updated 2012 Jun 28]. PMID: 20301670.
13. Da Paz J.A. Mowat-Wilson syndrome: neurological and molecular study in seven patients / J.A. da Paz, C.A. Kim, M. Goossens, I. Giurgea, M.J. Marques-Dias // Arq. Neuropsiquiatr. 2015 Jan; 73(1): 12-7. doi: 10.1590/0004-282X20140182.
14. Dagli A.I., Mueller J., Williams C.A. Angelman Syndrome // Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors // GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 1998 Sep 15 [updated 2015 May 14].PMID: 20301323.
15. De Leersnyder H. Smith-Magenis syndrome // Handb. Clin. Neurol. 2013; 111: 295-6. doi: 10.1016/B978-0-444-52891-9.00034-8.
16. Dentici M.L. Kabuki syndrome: clinical and molecular diagnosis in the first year of life / M.L. Dentici, A. Di Pede, F.R. Lepri et al. // Arch. Dis. Child. 2015 Feb; 100(2): 158-64. doi: 10.1136/archdischild-2013-305858.
17. Dornelles-Wawruk H. Complex phenotype associated with 17q21.31 microdeletion / H. Dornelles-Wawruk, A. Pic-Taylor, C. Rosenberg, A.C. Krepischi, H.P. Safatle, I. Ferrari, J.F. Mazzeu // Mol. Syndromol. 2013 Sep; 4(6): 297-301. doi: 10.1159/000354120.
18. Dziembowska M. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline / M. Dziembowska, D.I. Pretto, A. Janusz et al. // Am. J. Med. Genet. A. 2013 Aug; 161A(8): 1897-903. doi: 10.1002/ajmg.a.36023.
19. Grieco J.C. An open-label pilot trial of minocycline in children as a treatment for Angelman syndrome / J.C. Grieco, S.L. Ciarlone, M. Gieron-Korthals et al. // BMC Neurol. 2014 Dec 10; 14(1): 232. doi: 10.1186/s12883-014-0232-x.
20. Grigg-Damberger M., Ralls F. Treatment strategies for complex behavioral insomnia in children with neurodevelopmental disorders // Curr. Opin. Pulm. Med. 2013 Nov; 19(6): 616-25. doi: 10.1097/MCP.0b013e328365ab89.
21. Heald M. Discrimination training reduces high rate social approach behaviors in Angelman syndrome: proof of principle / M. Heald, D. Allen, D. Villa, C. Oliver // Res. Dev. Disabil. 2013 May; 34(5): 1794-803. doi: 10.1016/j.ridd.2013.02.012.
22. Huang H.S. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons / H.S. Huang, J.A. Allen, A.M. Mabb et al. // Nature. 2011 Dec 21; 481(7380): 185-9. doi: 10.1038/nature10726.
23. Jurecka A. Adenylosuccinate lyase deficiency / A. Jurecka, M. Zikanova, S. Kmoch, A. Tylki-Szymańska // J. Inherit. Metab. Dis. 2015 Mar; 38(2): 231-42. doi: 10.1007/s10545-014-9755-y.
24. Kleefstra T., Nillesen W.M., Yntema H.G. Kleefstra Syndrome // Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors // GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2010 Oct 5 [updated 2015 May 7]. PMID: 20945554.
25. Kolevzon A. Phelan-McDermid syndrome: a review of the lite–rature and practice parameters for medical assessment and monito–ring / A. Kolevzon, B. Angarita, L. Bush et al. // J. Neurodev. Disord. 2014; 6(1): 39. doi: 10.1186/1866-1955-6-39.
26. Koolen D.A. Mutations in the chromatin modifier gene –KANSL1 cause the 17q21.31 microdeletion syndrome / D.A. Koolen, J.M. Kramer, K. Neveling et al. // Nat. Genet. 2012 Apr 29; 44(6): 639-41. doi: 10.1038/ng.2262.;
27. Meng L. Towards a therapy for Angelman syndrome by targe–ting a long non-coding RNA / L. Meng, A.J. Ward, S. Chun, C.F. Bennett, A.L. Beaudet, F. Rigo // Nature. 2015 Feb 19; 518(7539): 409-12. doi: 10.1038/nature13975.
28. Motobayashi M. Neurodevelopmental features in 2q23.1 microdeletion syndrome: report of a new patient with intractable seizures and review of literature / M. Motobayashi, A. Nishimura-Tadaki, Y. Inaba et al. // Am. J. Med. Genet. A. 2012 Apr; 158A(4): 861-8. doi: 10.1002/ajmg.a.35235.
29. Mullegama S.V. Phenotypic and molecular convergence of 2q23.1 deletion syndrome with other neurodevelopmental syndromes associated with autism spectrum disorder / S.V. Mullegama, J.T. Alaimo, L. Chen, S.H. Elsea // Int. J. Mol. Sci. 2015 Apr 7; 16(4): 7627-43. doi: 10.3390/ijms16047627.
30. Mullegama S.V. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder / S.V. Mullegama, J.A. Rosenfeld, C. Orellana et al. // Eur. J. Hum. Genet. 2014 Jan; 22(1): 57-63. doi: 10.1038/ejhg.2013.67.
31. Paciorkowski A.R. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways / A.R. Paciorkowski, R.N. Traylor, J.A. Rosenfeld et al. // Neurogenetics. 2013 May; 14(2): 99-111. doi: 10.1007/s10048-013-0356-y.
32. Panda A.K., Kar S.K., Gopinath G. Angelman syndrome in three biological siblings: Focusing on the neuropsychiatric domain // J. Pediatr. Neurosci. 2013 Sep; 8(3): 213-6. doi: 10.4103/1817-1745.123674.
33. Pelc K. Epilepsy in Angelman syndrome / K. Pelc, S.G. Boyd, G. Cheron, B. Dan // Seizure. 2008 Apr; 17(3): 211-7. doi: http://dx.doi.org/10.1016/j.seizure.2007.08.004.
34. Pescosolido M.F. Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome / M.F. Pescosolido, D.M. Stein, M. Schmidt et al. // Ann. Neurol. 2014 Oct; 76(4): 581-93. doi: 10.1002/ana.24225.
35. Peters S.U. Double-blind therapeutic trial in Angelman syndrome using betaine and folic acid / S.U. Peters, L.M. Bird, V. Kimonis et al. // Am. J. Med. Genet. A. 2010 Aug; 152A(8): 1994-2001. doi: 10.1002/ajmg.a.33509.
36. Peters S.U., Byiers B.J., Symons F.J. Diurnal Salivary Cortisol and Regression Status in MECP2 Duplication Syndrome // J. Child. Neurol. 2015 May 21. pii: 0883073815585577.
37. Phelan K., Rogers R.C. Phelan-McDermid Syndrome // Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors // GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2005 May 11 [updated 2011 Aug 25].
38. Puffenberger E.G. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder / E.G. Puffenberger, R.N. Jinks, H. Wang et al. // Hum. Mutat. 2012 Dec; 33(12): 1639-46. doi: 10.1002/humu.22237.
39. Ramocki M.B., Tavyev Y.J., Peters S.U. The MECP2 duplication syndrome // Am. J. Med. Genet. A. 2010 May; 152A(5): 1079-88. doi: 10.1002/ajmg.a.33184.
40. Schwichtenberg A.J., Malow B.A. Melatonin Treatment in Children with Developmental Disabilities // Sleep Med. Clin. 2015 Jun; 10(2): 181-187. doi: 10.1016/j.jsmc.2015.02.008.
41. Stevenson R.E. Alpha-Thalassemia X-Linked Intellectual Disability Syndrome // Pagon R.A., Adam M.P., Ardin–ger H.H., Wallace S.E., Amemiya A., Bean L.J.H., Bird T.D., Dolan C.R., Fong C.T., Smith R.J.H., Stephens K., editors // GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. 2000 Jun 19 [updated 2014 Nov 06].PMID: 20301622.
42. Summers J.A. A combined behavioral/pharmacological treatment of sleep-wake schedule disorder in Angelman syndrome / J.A. Summers, P.S. Lynch, J.C. Harris, J.C. Burke, D.B. Allison, L. Sandler // J. Dev. Behav. Pediatr. 1992 Aug; 13(4): 284-7. PMID: 1506469.
43. Tan W.H. If not Angelman, what is it? A review of Angelman-like syndromes / W.H. Tan, L.M. Bird, R.L. Thibert, C.A. Williams // Am. J. Med. Genet. A. 2014 Apr; 164A(4): 975-92. doi: 10.1002/ajmg.a.36416.
44. Thibert R.L. Epilepsy in Angelman syndrome: a questionnaire-based assessment of the natural history and current treatment options / R.L. Thibert, K.D. Conant, E.K. Braun et al. // Epilepsia. 2009 Nov; 50(11): 2369-76. doi: 10.1111/j.1528-1167.2009.02108.x.
45. Williams C.A., Driscoll D.J., Dagli A.I. Clinical and genetic aspects of Angelman syndrome // Genet. Med. 2010 Jul; 12(7): 385-95. doi: 10.1097/GIM.0b013e3181def138.

/83.jpg)