Статья опубликована на с. 139-142
Введение
Врожденная аплазия кожи — это порок развития, характеризующийся очаговым дефектом кожи, который может сочетаться с аплазией глубже расположенных тканей, в частности с аплазией костей и различными формами дизрафий — незакрытием эмбриональных щелей.
Данные о частоте данного врожденного порока широко варьируют и составляют 1,1 на 1000 новорожденных, по данным латиноамериканских исследователей [3], и 1 к 5000 новорожденных, по данным других авторов [5].
Аплазия кожи, кости может быть результатом фетопатии, возникающей при воздействии на плод различных неблагоприятных факторов: вирусной, бактериальной инфекции, интоксикации, малых доз радиации, приема тимазола или метимазола во время беременности [1].
Возможны наследственные формы с аутосомно-доминантным или рецессивным типом наследования. Описаны случаи хромосомных аномалий (трисомия по 13-й паре, делеция короткого плеча 4-й хромосомы). Описаны несколько видов аплазии кожи в зависимости от локализации, в том числе наиболее часто встречающийся синдром Андерсона — Нови (Кемпбелла синдром) — аплазия кожи черепа, которая характеризуется дефектом кожи (до 10 см в диаметре) мембранозного или буллезного характера, локализующимся обычно в теменной области, реже в затылочной и задней аурикулярной области, сочетающимся с дефектом костей черепа. Очаг аплазии одиночный в 70 % случаев, однако возможно наличие 2–3 очагов поражения, реже более. Мембранозные очаги обычно округлые, лишены кожи, подлежащих тканей, включая костную, и покрыты тонкой эпителиальной мембраной. Гистологически в области мембранозного дефекта обнаруживают компактный коллаген с уменьшением количества эластических волокон, придатки кожи отсутствуют. Возможны признаки воспаления, некроза.
I. Frieden (1986) предложила классификацию врожденных аплазий кожи, разделив их на 8 групп:
I группа — врожденные аплазии кожи волосистой части головы без аномалий или исключительно с изолированными аномалиями.
II–VIII группы — врожденные аплазии кожи, сочетающиеся с рядом других пороков и аномалий в составе синдромов развития [5]. Например, аплазия кожи волосистой части головы является одним из проявлений синдрома Йохансона — Близзара, включающего такие аномалии, как аплазия крыльев носа, микроцефалия и задержка умственного развития, отсутствие зачатков постоянных зубов, глухота, карликовый рост, дефицит внешнесекреторной функции поджелудочной железы, первичный гипотиреоз, который приводит к истончению, разрежению волос на голове и их гипопигментации [2].
Патоморфологически аплазия кожи во всех описанных группах может ограничиваться только отсутствием ее придатков. Эпидермис обычно полностью отсутствует, а если есть дерма, то она лишена полноценных эластических волокон и содержит компактный коллаген. Дефект может захватывать подкожную жировую клетчатку, череп, твердую мозговую оболочку и иногда подлежащий участок мозга c образованием дорсального энцефалоцеле.
Врожденная аплазия кожи черепа может сочетаться с врожденной гидроцефалией, гемангиомами, поликистозом почек, менингоцеле, расщеплением неба, микрофтальмией, буллезным эпидермолизом. Возможны осложнения в виде присоединения вторичной инфекции, менингита при сообщении кожного дефекта с мозговыми оболочками, кровотечения из сагиттального синуса.
Дефекты кожи могут быстро заживать с образованием атрофического или келоидного рубца. Рецидивирующее образование в очагах корок может замедлять их заживление на месяцы или годы. Даже обширные поверхностные аплазии кожи заживают, но с видимыми белыми участками — пленками, иногда рубцово измененными.
Смертность у 20 % детей с аплазией кожи волосистой части головы связана с менингитом, кровотечениями, эрозиями сагиттального синуса и дыхательной недостаточностью. Аплазия кожи может сочетаться с аномалией вен, обусловливающей высокий риск возникновения кровотечения. В редких случаях атрофия подлежащих участков мозга может приводить к спастическому параличу и задержке умственного развития [2, 5].
В некоторых случаях участки аплазии в момент рождения имеют вид пузырей, которые вскоре вскрываются. Пузыри сочетаются с очагами аплазии в виде раневой поверхности и напоминают буллезный эпидермолиз.
Прогноз зависит от локализации аплазии, глубины и степени поражения подлежащих тканей и органов. Лечение направлено на предупреждение менингоэнцефалита и заживление эрозивно-язвенного дефекта, выявление и коррекцию других врожденных пороков. Зона аплазии при заживлении имеет вид косметического безволосого мембранозного дефекта белого цвета [2].
Цель работы: учитывая редкую встречаемость врожденной аплазии кожи в практике, приводим описание врожденного синдрома Андерсона — Нови (Кемпбелла синдром), который характеризуется дефектом кожи, локализующимся в теменной области, и сочетается с дефектом костей черепа.
Материалы и методы
В работе использован клинический метод наблюдения; ультразвуковой метод исследования: динамическое нейросонографическое исследование выполняли через большой родничок секторальными датчиками 5 МГц (аппарат Sonoline 40) путем последовательного получения 10 стандартных сечений в коронарной и сагиттальной плоскостях по общепринятой методике.
Ребенок родился путем операции кесарева сечения с оценкой по шкале Апгар 7–8 баллов, с массой тела 3090 г, ростом 50 см, окружностью головы 34 см, окружностью груди 34 см в сроке гестации 39 недель от I беременности I родов у женщины 24 лет. Женщина имеет высшее образование, работает, замужем. Вредных привычек, профессиональных вредностей нет. В соматическом анамнезе у женщины имеет место пиелонефрит с детства, гинекологический анамнез — эрозия шейки матки. В течение беременности дважды болела ОРЗ с повышением температуры. Фенотипически данных о синдромальной патологии у женщины нет. Мужу 24 года, здоров, профессиональных вредностей, вредных привычек нет. Генеалогический анамнез семьи не отягощен.
Результаты
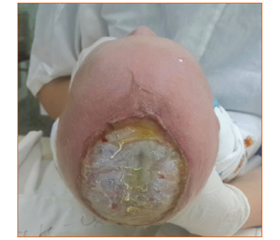
Ребенок переведен из родильного дома города Харькова в Харьковский городской перинатальный центр в связи с выявленным дефектом в области теменных костей черепа сразу после рождения. При поступлении мальчик активный, рефлексы периода новорожденности вызываются в полном объеме. При осмотре головы по ходу сагиттального шва определяется правильной формы дефект кожи размером 10 × 10 см и таких же размеров дефект двух теменных костей. Просматривается твердая мозговая оболочка, верхний сагиттальный синус и вены, впадающие в него (рис. 1).
Кожа туловища и слизистые розовые, чистые. В легких — везикулярное дыхание. Тоны сердца ритмичные, частота сердечных сокращений 146 в 1 мин, в пятой точке выслушивается нежный систолический шум. Живот мягкий, безболезненный. Печень определяется на 1 см ниже края реберной дуги, селезенка не пальпируется.
Ребенок обследован: клинический и биохимический анализы крови, клинический анализ мочи, ЭКГ, УЗИ головного мозга, сердца, органов брюшной полости, МРТ головного мозга, консультирован специалистами.
При проведении цитогенетического исследования № 1007-15 методом культивирования лимфоцитов периферической крови методом С-G окрашивания выделен кариотип 46 ХY, 1 % нестабильности. Можно предположить, что в основе порока лежит нарушение развития эктодермального и мезодермального листков, вследствие чего произошло нарушение развития костей черепа и кожных покровов головы. Дефект возник при одновременном действии некоторых факторов внешней среды на фоне генетической предрасположенности (полигенное наследование).
При МРТ-исследовании: гипоксически — ишемическое поражение белого вещества головного мозга, небольшой участок энцефаломаляции в корковых отделах левой теменной доли.
По данным нейросонографии с допплерометрией сосудов головного мозга выявлена перивентрикулярная ишемия первой степени, дистонический характер мозгового кровотока (повышение IR в левой передней мозговой артерии). По данным допплерэхокардиографии — наличие аномальной хорды в полости левого желудочка, функционирующее овальное окно 2 мм. При проведении ультразвукового исследования брюшной полости патологии не выявлено.
При осмотре детским неврологом выявлено: голова округлой формы, отсутствует кожа и две теменные кости черепа. Глазные щели D = S, лицо симметрично. Сосет, глотает. Объем спонтанной моторики снижен. Мышечный тонус дистоничен. Сухожильные рефлексы живые. Рефлексы периода новорожденности снижены в группе спинальных автоматизмов.
Рекомендации нейрохирурга: у ребенка с врожденной патологией покровов головы и костей черепа совместно с комбустиологами в дальнейшем будет решаться вопрос о пластическом закрытии кожного дефекта.
Обсуждение
Во время пребывания в стационаре ребенок наблюдался комбустиологом, нейрохирургом, хирургом. В динамике имело место уменьшение диаметра раневой поверхности, формирование участков грануляции по центру и по периферии. Твердая мозговая оболочка покрывалась фибрином. Ребенок 2 раза в неделю по настоящее время получает перевязки с гроссалиндом (защищает твердую мозговую оболочку от пересыхания), мазью левомеколь и мазью сульфаргин. Проведен курс антибактериальной, ноотропной терапии. В настоящий момент возраст ребенка составляет 2 месяца. Площадь поверхности дефекта 2,5 × 5 см, покрыта грануляциями по центру и по периферии (рис. 2, 3).
Выводы
У доношенного ребенка диагностирована редкая врожденная патология — cиндром Андерсона — Нови (Кемпбелла синдром), которая может быть результатом фетопатии, возникающей при воздействии различных неблагоприятных факторов, влияющих на плод. Врожденная патология сопровождалась гипоксическими изменениями белого вещества головного мозга с небольшим участком энцефаломаляции в корковом отделе левой теменной кости, что подтверждено данными МРТ-исследования.
Лечебные мероприятия направлены на предупреждение менингоэнцефалита и развития сепсиса, заживление раневого дефекта, улучшение кровообращения и обменных процессов в головном мозге.
При благоприятном исходе и заполнении обширных дефектов в области теменных костей в последующем возможно применение методов пластической хирургии.
Авторы заявляют об отсутствии конфликта интересов.

/141.jpg)