
Международный неврологический журнал №4 (106), 2019
Вернуться к номеру
Спинальные мышечные атрофии у детей: эпидемиология, фенотипические особенности и современные возможности ранней клинико-инструментальной диагностики
Авторы: Шаймурзин М.Р.
Чеченский государственный университет, г. Грозный, Россия
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
У статті розглянуті особливості клінічних проявів з позиції фенотипових особливостей проксимальних спінальних м’язових атрофiй (СМА). Надані міжнародні верифіковані шкали оцінки функціональних можливостей дітей зi СМА, рекомендовані для застосування в практичній охороні здоров’я для вірогідної оцінки динаміки змін клінічних проявів захворювання. Наведений адаптований мінімальний клінічний патерн для вірогідного диференціювання форми СМА в межах однієї нозологiчної категорії. Обговорюються міждисциплінарні заходи з ранньої діагностики СМА. Акцент зроблено на тривалому клініко-електроміографічному моніторингу, що дозволяє припустити прогресування захворювання на ранніх стадiях. Виявлено основні причини помилок в діагностуванні СМА у дітей. Розглянуто проблему реабілітаційних програм у віковому аспекті.
В статье рассмотрены особенности клинических проявлений с позиции фенотипических особенностей проксимальных спинальных мышечных атрофий (СМА). Представлены международные верифицированные шкалы оценки функциональных возможностей детей со СМА, рекомендованные для применения в практическом здравоохранении в целях достоверной оценки динамики изменений клинических проявлений заболевания. Представлен адаптированный клинический паттерн для достоверной дифференцировки формы СМА в пределах одной нозологической категории. Обсуждается междисциплинарный подход к ранней диагностике СМА. Акцент сделан на длительном клинико-электромиографическом мониторинге, позволяющем достоверно оценить прогрессирование заболевания на ранних сроках. Выявлены основные причины клинических ошибок в диагностике СМА у детей. Рассмотрена проблема реабилитационных программ в возрастном аспекте.
The article describes the features of clinical manifestations from the position of phenotypic features of proximal spinal muscular atrophies (SMA). The paper deals with the international verified scales for performance assessment in children with SMA, recommended for use in practical health care in order to reliably assess the dynamics of clinical manifestations of the disease. An adapted clinical pattern is presented for reliable differentiation of the SMA form within the same nosological category. A multidisciplinary approach to the early diagnosis of SMA is discussed. The emphasis is placed on long-term clinical electromyographic monitoring, which allows reliably assess the progression of the disease at the early stages. The main causes of clinical errors in the diagnosis of SMA in children are identified. The problem of rehabilitation programs in the context of age is considered.
спінальні м’язовi атрофії; клінічні прояви; функціональні та лабораторні методи дослідження СМА; діагностичні помилки; можливості терапії
спинальные мышечные атрофии; клинические проявления; функциональные и лабораторные методы исследования СМА; диагностические ошибки; возможности терапии
spinal muscular atrophies; clinical manifestations; functional and laboratory methods for SMA study; diagnostic errors; treatment options
Наследственные нейромышечные заболевания (НМЗ) — генетически гетерогенная группа патологий моторной интеграции, отличительным свойством которой является клинический полиморфизм с сопутствующими стойкими и достаточно часто тяжелыми двигательными нарушениями, со значительным снижением функциональных возможностей больного, ограничением возможностей самообслуживания и развитием инвалидизации. Частота НМЗ составляет 7,2 случая на 100 000 человек в общей популяции [1]. Но в этом сегменте лидирующие позиции занимают спинальные мышечные атрофии (СМА), характеризующиеся прогрессирующей нейродегенерацией структур и гибелью мотонейронов передних рогов спинного мозга [8]. Повышенное внимание к проблемам СМА также обусловлено неуклонным увеличением числа пациентов с данной патологией в последние десятилетия [6, 19]. Основными причинами увеличения частоты распространенности СМА являются как истинное нарастание заболеваемости, так и медико–социальные факторы: возросшее количество выживших незрелых новорожденных, рожденных с критически низкой массой тела (< 500 г), что привело к появлению нового поколения страдающих генными мутациями с позиции особенностей онтогенеза, а также улучшение возможностей молекулярно–генетической верификации [2].
В структуре СМА детского возраста доминируют изолированные проксимальные СМА с аутосомно–рецессивным типом наследования (I, II, III, IV), со спе–цификацией гена выживаемости мотонейронов SMN1 (survival motor neuron), локализующегося на длинном плече хромосомы 5 (5q13) [6]. Распространенность проксимальных СМА составляет 5,5 на 100 тыс. населения, среди новорожденных — 1 на 6–10 тыс. [11]. Отмечается значительное преобладание пациентов мужского пола, особенно среди лиц с началом заболевания от 37 месяцев до 18 лет [14]. Частота встречаемости гетерозиготного носительства достигает 1 на 40–60 человек [4]. Определение связей между маркерами в хромосомной области 5q11.2–13.3 для СМА I–III типов и открытие необходимых генов–кандидатов стало основой для классификации по фенотипической изменчивости (табл. 1).
/74-1.jpg)
В США группа СМА представляет собой второе по распространенности аутосомно–рецессивное расстройство после кистозного фиброза [15]. Острая форма заболевания в младенческом возрасте (I тип) встречается примерно у одного из 10 000 младенцев, рожденных живыми; хронические формы (II и III типы) встречаются у одного из 24 000 младенцев [10]. Частота генной мутации в Великобритании оценивается как 1 к 160, а частота появления носителя (гетерозиготного) — как 1 к 60–80 [7]. Имеются данные о высоких показателях частоты заболеваемости у этнической группы караимов в Израиле (1 к 20) [15]. По данным ВОЗ, СМА I типа составляет около одной четвертой всех случаев. СМА II типа является наиболее распространенной и составляет половину всех случаев. СМА III типа составляет одну четвертую всех случаев [14]. Исходя из подобных данных, можно констатировать, что истинная частота проксимальных СМА более высокая, так как часть случаев установки ошибочного диагноза или внезапной смерти остается недиагностированной.
Согласно фактическим данным, показатели заболеваемости и смертности зависят от возраста дебюта первых симптомов заболевания [2, 3]. Для заболевания в раннем возрасте характерны более высокие показатели смертности [9, 12]. Например, пациенты с СМА I типа после манифестации заболевания в среднем живут 7 месяцев, при этом смертность по достижении 18–месячного возраста составляет 95 % [10]. При внутри–утробном заболевании средний возраст смерти составляет 6 месяцев [4]. У младенцев и детей с СМА I типа самой распространенной причиной смерти являются респираторные инфекции [12].
Актуальнейшим аспектом поднимаемых проблем является заострение внимания на роли своевременной диагностики и обращаемости к профильным специалистам. В настоящее время на вооружении у врача достаточно широкий спектр диагностических возможностей, но, к сожалению, частота диагностических ошибок и поздней верификации инициальных симптомов заболевания остается высокой [2, 17]. Зачастую дети длительно наблюдаются по месту жительства по поводу перинатальной энцефалопатии c синдромом двигательных нарушений или мышечной гипотонии, дисплазии тазобедренных суставов, плоскостопия или вальгусной деформации стоп. Подобная дефектура также определяется недостаточной осведомленностью практических врачей первичного звена, в свою очередь обусловленной и тем, что нет определенного разработанного адаптированного клинического паттерна, позволяющего заподозрить ранние проявления СМА и выделить ведущие симптомы, характеризующие форму заболевания в пределах одной нозологической категории. Адекватная формулировка диагноза СМА согласно международной классификации на первичных этапах встречается в единичных случаях. Нередко диагноз ограничивается «синдромом вялого ребенка» или нейромышечным заболеванием. За последнее десятилетие клинико–диагностической работы уточнение формы СМА происходило в 75 % случаев. В ряде клинических ситуаций возникала необходимость кардинально менять диагноз со СМА на другой вид нейромышечного заболевания, включая структурную миопатию, митохондриопатию, аномалию спинного/головного мозга, или наоборот. В остальных случаях происходило уточнение формы в пределах одной категории. Таким образом, ошибки в диагностике нейромышечных заболеваний у детей можно объяснить несвоевременным осмотром, отсутствием динамического наблюдения, неверной дифференциацией нормального и девиантного неврологического статуса, недостаточным знанием неврологами первичного звена семиотики и синдромологии СМА детского возраста, ошибочной трактовкой данных анамнеза. Анализируя сложившуюся ситуацию, сегодня необходимо акцентировать внимание на базисных клинических признаках и семиотике двигательных нарушений различных вариантов СМА, целесообразно разработать и внедрить паттерны ранней диагностики СМА на период обращения ребенка и принять их на вооружение врачами первичного звена.
В некоторых публикациях [4, 19] указывается, что у ряда пациентов признаки проксимальных СМА проявляются на стыке классифицированных типов. Кроме этих определяющих критериев, современные клинические признаки различных вариантов СМА включают следующие:
1. СМА I типа — острое заболевание у младенцев, тяжелая форма (болезнь Верднига — Гоффмана), описана G. Werdnig в 1891 году. К данному типу относятся до 30 % всех случаев СМА [7]. Данный тип заболевания проявляется в возрасте от 0 до 6 месяцев [9]. В 95 % случаев он проявляется в возрасте до 3 месяцев. Тип наследования — аутосомно–рецессивный. Характерными признаками СМА I типа являются стремительное прогрессирование симптомов, стойкий цианоз при рождении, глубокая слабость и выраженная диффузная мышечная гипотония; бульбарные нарушения; атрофия и фасцикуляции языка, нарастание мышечной слабости в конечностях, приводящей к резкому выраженному ограничению активных движений [1–4, 11—13]. Средняя продолжительность жизни ребенка со СМА I типа составляет в среднем 5,9 месяца; 95 % пациентов не доживают до возраста 2 лет. За редким исключением у преобладающего процента пациентов отсутствуют навыки сидения без поддержки. Витальный прогноз связан с дыхательными нарушениями, преимущественно в виде аспирационной пневмонии [12]. Ряд авторов [9–11] выделяют среди СМА I типа наиболее тяжелый вариант течения — врожденную форму с послеродовой асфиксией. На 30–34–й неделе беременности отмечают малоподвижность плода. Ведущим клиническим признаком является периферическая тетраплегия с контрактурами, нарушением глотания и дыхания: без технического обеспечения дыхательной поддержки ребенок может погибнуть. Такой вариант СМА классифицируют как СМА 0 типа.
2. СМА II типа, промежуточный вариант, предложенный V. Dubowitz [3]. Наиболее распространенный вариант проксимальной СМА (45 % случаев), нередко ассоциированной с другими изолированными проксимальными формами СМА [4, 11, 14]. Наследуется по аутосомно–рецессивному типу. Как правило, статомоторное развитие детей в первые 6 месяцев жизни проходит без отклонений. В ряде случаев регресс двигательных навыков проявляется до достижения ими 18–месячного возраста. Клиническая симптоматика представлена слабостью мышц грудного пояса и проксимальных мышц, арефлексией, фасцикулярным, эмоционально индуцируемым тремором верхних конечностей, нейрогенными контрактурами суставов нижних и верхних конечностей, часто в сочетании с подвывихами в тазобедренных суставах, кифосколиозом, гиперлордозом, слабостью межреберных мышц, поверхностным дыханием с ослаблением пассажа секрета из бронхиол [1–7, 11, 13, 18]. За редким исключением у детей со СМА отсутствуют навыки самостоятельной ходьбы [7]. Уровень креатинкиназы может превышать норму не более чем в пять раз. Продолжительность жизни варьирует от 2 до 20–30 лет. Причиной большинства ранних смертей являются респираторные инфекции [12]. Благоприятный прогноз — у детей, способных стоять у опоры или с помощью ортопедических приспособлений [13].
3. СМА III типа — юношеская форма (болезнь Кугельберга — Веландера), описанная E. Kugelberg и L. Welander в 1956 году. К данному типу относится около 18 % всех случаев заболевания. Данная доброкачественная форма заболевания проявляется у пациентов в возрасте старше 18 месяцев. Наиболее часто инициальные признаки болезни появляются в возрастном диапазоне 2–7 лет в проксимальных группах мышц нижних конечностей и тазового пояса [6]. Больные предъявляют жалобы на затруднения при ходьбе, беге, подъеме или спуске, приседании [1–6, 14, 17]. Нередко отмечаются псевдогипертрофии мышц нижних конечностей, миалгии по типу крампи. В этот период клинико–неврологические проявления имеют определенную идентичность с клинической симптоматикой при дистрофинопатии Беккера. Слабость мышц симметричная, представлена преимущественно в проксимальных отделах, более выражена в нижних конечностях. Характерной является гипермобильность суставов; по мере прогрессирования заболевания формируется поясничный гиперлордоз. Деформации скелета встречаются редко. Позже могут присоединяться дисфагия и дизартрия. Болезнь прогрессирует медленно, и в большинстве случаев можно ожидать нормальную продолжительность жизни. Уровень креатинкиназы чаще в норме, повышение описывают при наличии вторичных миопатических изменений. Пациенты длительно сохраняют способность к самостоятельному передвижению, нередко осложняющемуся «немыми» переломами, локализованными в местах соединения сухожилия с костным отростком. В литературе встречается разделение СМА III типа на два подтипа, IIIa и IIIb, в зависимости от времени начала заболевания: до трех лет и после трех лет соответственно [5].
4. СМА IV типа. Дебют заболевания — в возрастном диапазоне от 10 до 30 лет. Характерной является стертость клинических признаков в виде незначительного ограничения функции передвижения, затруднений при беге, приседаниях, прыжках, умеренной гипотрофии мышц бедер и тазового пояса [4–6]. Для СМА IV типа респираторные и кишечно–желудочные осложнения не специфичны [12]. Течение заболевания чаще стационарное.
Таким образом, минимальный, но достоверный спектр первичного обследования фенотипических вариантов СМА должен базироваться на совокупности прогрессирующих клинических признаков: 1) СМА I типа (болезнь Верднига — Гоффмана). Дебют — 0–6 мес. Ослабленный контроль головы, слабый плач, снижены кашлевой, глотательный рефлексы, прием пищи и слюноотделения нарушаются до 1 года, атрофия и фасцикуляции языка, выраженная слабость и гипотония конечностей и туловища, поверхностное (диафрагмальное) дыхание, деформация грудной клетки; 2) СМА II типа. Дебют — 6–18 мес. Регресс двигательных навыков, предшествующий нормальному моторному развитию, когнитивные нарушения не характерны, недостаточная прибавка веса, слабость и гипотрофия мышц плечевого пояса и верхней части туловища, интенционный тремор, прогрессирующее развитие кифосколиоза и контрактур крупных суставов, в положении сидя представлен кифоз; 3) СМА III типа (болезнь Кугельберга — Веландера). Дебют после 18 месяцев. Способность к самостоятельному передвижению. Гипотрофия и слабость мышц тазового пояса, бедер и плечевого пояса, боли в мышцах по типу крампи. Некоторые пациенты теряют способность ходить в детстве (до 30 % случаев). В 80 % случаев наблюдается сколиоз. Распространены боли в мышцах; 4) СМА IV типа. Дебют — от 10 до 30 лет. Стертость клинических признаков в виде незначительного ограничения функции передвижения, затруднений при беге, приседаниях, прыжках, умеренной гипотрофии мышц бедер и тазового пояса. Течение заболевания мягкое.
Сегодня приоритетным направлением реабилитации является социально–педагогическая помощь ребенку, безусловно, играющая немаловажную роль в улучшении качества жизни и социальной адаптации. К сожалению, основные положения реабилитации в социальных центрах не включают последующее уточнение и пересмотр диагноза и терапии. Как свидетельствует практика, без медикаментозной поддержки подобный подход является прогностически неблагоприятным мероприятием [2, 3, 13]. Как показал анализ литературы, число специализированных медицинских центров реабилитации детей с наследственной нейромышечной патологией исчисляется единицами. В то же время организация специализированных коек для детей с нейромышечной патологией в общепрофильных центрах нейрореабилитации является эффективным подходом, поскольку осуществляется специализированная помощь и компетентный подход к терапии с оказанием долговременной квалифицированной медицинской помощи, включая применение телекоммуникационных технологий и Интернета (онлайн–консультации, онлайн–конференции и прочее). Подобный подход обеспечивает преемственность между социальными и специализированными клиническими центрами, формируя междисциплинарную связь по смежным проблемам.
Наряду с этим наследственная нейромышечная патология приобретает все более отчетливое медико–социальное звучание и представляет собой одну из важных проблем практического здравоохранения ввиду постоянного увеличения частоты ее встречаемости. Это послужило основанием для формирования 4 специализированных коек для детей с нейромышечной патологией на базе ГБУ «Республиканская детская клиническая больница им. Е.П. Глинки» (Чеченская Республика) и в последующем — создания структурного подразделения для детей с наследственной нейромышечной патологией в структуре ГБУ «Республиканский детский реабилитационный центр» (Чеченская Республика). За период 2000–2016 гг. в реестре базы данных ГБУ зарегистрировано 107 детей со спинальными амиотрофиями, в том числе СМА I типа — болезнью Верднига — Гоффмана (n = 8; 7,5 %), II типа — промежуточного (n = 60; 56,1 %), III типа — болезнью Кугельберга — Веландера (n = 21; 19,6 %), IV типа (n = 12; 11,2 %).
Согласно международным стандартам, диагностический алгоритм включает медико–генетическое исследование, оценку функциональных возможностей пациента с учетом рекомендованных международными экспертами верифицированных шкал RHS (модифицированная шкала Хаммерсмит), RULM (модифицированный модуль оценки манипулятивной функции), функциональные методы исследования, включая ЭНМГ, спирометрию, ЭКГ, радиологические методы (денситометрия, рентгенография, магнитно–резонансная томография), биохимическое исследование (КФК, лактат, витамин D3, ионизированный кальций).
Согласно определению, RHS — это оценка физических возможностей пациентов со СMA II и III типов с ограниченными функциями передвижения для мониторинга функции сидения, стояния, ходьбы, диапазона движений и силы в конечностях, мышцах туловища и шеи. Применяется для детей 5 лет и старше. RHS является порядковой шкалой, состоящей из 33 элементов в диапазоне от 0 до 3 баллов, где 0 обозначает наименьший уровень способности/функции, развивающийся до наивысшего уровня способности успешно выполнить задание — 2 балла. Максимально достижимая оценка — 69. Тем не менее шкала RHS недостаточно эффективно отражает прогрессирующую мышечную слабость в стадии стойких тяжелых двигательных нарушений с низким уровнем функциональных возможностей. Для данной категории детей со СМА применяется международная шкала RULM (Revised upper limb module for spinal muscular atrophy). В состав шкалы RULM входит 20 тестов, таких как перенос рук с колен на стол, сбор мелких предметов, нажатие на кнопки, разрывание бумаги, открытие пластикового контейнера, перенос рук выше плеч и поднятие предметов разного веса на разную высоту.
Основополагающим среди критериев достоверной диагностики является поиск генетической мутации, что рассматривается в качестве золотого стандарта [1, 9].
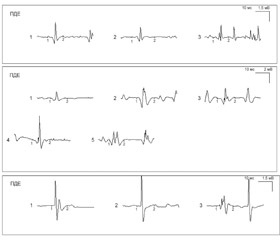
Наряду с современными методами исследования (включая магнитно–резонансную томографию аксиальных мышц и туловища, позитронно–эмиссионную томографию, нейросонографическое исследование нейромышечного аппарата, гистохимическое исследование мышечного биоптата и пр.), к сожалению, не всегда доступными для широких слоев детского населения, в последние годы все больше внимания уделяется нейрофизиологическим маркерам, которые позволяют определить уровень поражения моторной интеграции на различных стадиях заболевания и оценить эффективность лечения. Изучая настоящий аспект проблемы применительно к СМА детского возраста, представляется целесообразным уделить особое внимание рассмотрению и идентификации маркеров раннего выявления прогрессирования заболевания. Во–первых, их знание необходимо для прогнозирования развития и течения заболевания, во–вторых — для определения контингента лиц, нуждающихся в долговременном целенаправленном дообследовании, и в–третьих — для установления первоочередности и характера назначаемой терапии. При установлении диагноза СМА и ее вариантов следует учитывать особенности клинических проявлений, результаты медико–генетического обследования, а также показатели электронейромиографии. В то же время существенным моментом, во многом определяющим раннюю диагностику патологических симптомов, является мониторирование динамики изменений клинических проявлений заболевания, подтвержденных цифровыми данными международных оценочных шкал и нейрофизиологическими показателями с помощью компьютерной электронейромиографии. Нами разработана и внедрена авторская методика ЭМГ–мониторинга (патент № 106146), основными моментами которой являются ежеквартально выполняемое ЭНМГ–исследование (рис. 1, 2), оценка интервального изменения миографических параметров, сопоставление с данными клинико–неврологического статуса, выделение специфических нейрофизиологических маркеров, позволяющих субклинически выявить утяжеление патологии и своевременно отреагировать терапевтическими мероприятиями.
/78-1.jpg)
/78-2.jpg)
В свою очередь, одним из патогномоничных клинических симптомов у детей со СМА являются легочные заболевания, наиболее актуальные для СМА I типа и в меньшей степени представленные у пациентов со СМА II типа [6, 14, 15]. Без обеспечения вспомогательной искусственной вентиляции легких младенцы, не умеющие сидеть, обычно погибают в возрасте до 2 лет. Причиной легочных нарушений является сочетание инспираторной и экспираторной мышечной слабости с большим вовлечением экспираторных и межреберных мышц (рис. 3).
/79-1.jpg)
Также актуальной и вместе с тем далеко не решенной проблемой является раннее выявление, предупреждение и терапия остеопороза/остепении [2, 3]. Для решения этого вопроса показаны денситометрия и выполнение биохимического исследования ионизированного кальция и витамина D3, уровень концентрации которого является основой селективного лечения (от коррекции диетой и ЛФК до введения бисфосфонатов) (рис. 4).
/79-2.jpg)
Таким образом, актуальность поднимаемой темы может быть резюмирована следующим образом: 1) именно поздняя диагностика влияет на течение заболевания; 2) несмотря на перспективность разрабатываемых подходов и успех в доклинических испытаниях этиотропной генной терапии, их разработка и внедрение в практическое здравоохранение находятся лишь на начальной стадии. Далеко не все из них показывают достаточно высокую эффективность, окончательно не разработана наиболее эффективная система доставки генотерапевтических препаратов, более того, зачастую невозможно применять универсальные подходы для терапии СМА с различными фенотипами, что сопряжено со значительным сужением целевой группы пациентов. С одной стороны, современные подходы к лечению основываются лишь на симптоматической коррекции двигательных нарушений, возникающих по мере прогредиенции процесса, а не их предупреждении или замедлении. С другой — не изучен этапный выбор терапии в целом. Актуален комплексный подход к оценке лечения двигательных нарушений как с клинических позиций, так и с применением ЭМГ на различных этапах заболевания, что обеспечит эффективный подбор этапоспецифичной терапии, направленной на замедление патологического процесса; 3) вопросы реабилитационной терапии пациентов после достижения ими совершеннолетия представлены чрезвычайно фрагментарно, что прежде всего обусловлено привязкой СМА к диагнозу детского возраста, поскольку медицинское сообщество продолжает рассматривать это заболевание в сегменте педиатрического диагноза. Более того, программа резидентуры для врачей, работающих со взрослыми, не содержит курс ведения пациентов со СМА. Поэтому для комплексной координированной помощи, направленной на профилактику осложнений и активизацию компенсаторно–приспособительных функций нейромышечного аппарата, необходима модификация предоставляемого реабилитационного лечения, целостное и глубокое изучение и решение практических задач, направленных на предупреждение инвалидизации на протяжении всей жизни пациента, что в конечном счете может улучшить качество его жизни и, с учетом потребностей больного, открыть доступ к его образовательным и профессиональным возможностям.
Конфликт интересов. Автор заявляет об отсутствии какого–либо конфликта интересов при подготовке данной статьи.
1. Гузева В.И. и др. Детская неврология: Клинические рекомендации. М.: СИМК, 2015. 332 с.
2. Евтушенко С.К., Шаймурзин М.Р. Нейромышечные заболевания у детей: проблемы ранней диагностики и современной медицинской и социальной реабилитации (научный обзор и собственные наблюдения). Межд. невр. журн. 2013. Т. 5, № 59. С. 13–33.
3. Евтушенко С.К. и др. Нейромышечные заболевания у детей. Донецк: Ноулидж, 2014. 218 с.
4. Петрухин А.С., Бобылова М.Ю. Детская неврология. М.: ГЭОТАР–Медиа, 2018. Т. 2. 555 с.
5. Соколова М.Г. и др. Спинальная мышечная атрофия у детей: этиология, патогенез, диагностика и принципы лечения. Вестник СЗГМУ. 2013. Т. 5, № 4. С. 108–114.
6. Яхно Н.Н., Парфенов В.А., Дамулин И.В. Нервные болезни. Общая неврология. М.: МИА, 2014. 256 с.
7. Arnold W.D. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve. 2015. Vol. 51, № 2. P. 157–67.
8. Arnold E.S. Spinal muscular atrophy. Handb. Clin. Neurol. 2018. Vol. 148. P. 591–601.
9. Carré A. Review of Spinal Muscular Atrophy (SMA) for Prenatal and Pediatric Genetic Counselors. J. Genet. Couns. 2016. Vol. 25, № 1. P. 32–43.
10. De Sanctis R. [et al.]. Clinical phenotypes and trajectories of disease progression in type 1 spinal muscular atrophy. Neuromuscular Disorders. 2018. Vol. 28. P. 24–28.
11. Darras B.T. Spinal muscular atrophies. Pediatr. Clin. North Am. 2015. Vol. 62, № 3. P. 743–66.
12. Finkel R.S. [et al.]. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018. Vol. 28, № 3. P. 197–207.
13. García–Salido A. [et al.]. Palliative care in children with spinal muscular atrophy type I: What do they need? Palliat Support Care. 2015. Vol. 13, № 2. P. 313–316.
14. Mercuri E. [et al.]. Patterns of disease progression in type 2 and 3 SMA: Implications for clinical. Neuromuscular Disorders. 2016. Vol. 26, № 2. P. 126–131.
15. Kraszewski J.N. [et al.]. Pilot study of population–based newborn screening for spinal muscular atrophy in New York state. Genet. Med. 2018. Vol. 20, № 6. P. 608–613.
16. Higashihara M. [et al.]. Quantitative Analysis of Surface Electromyography for Pediatric Neuromuscular Disorders. Muscle Nerve. 2018. Vol. 58, № 6. P. 824–827.
17. Sampaio H., Wilcken B., Farrar M. Screening for spinal muscular atrophy. Med. J. Aust. 2018. Vol. 209, № 4. P. 147–148.
18. Shababi M., Lorson C.L., Rudnik–Schöneborn S.S. Spinal muscular atrophy: a motor neuron disorder or a multi–organ disease? J. Anat. 2014. Vol. 224, № 1. P. 15–28.
19. Vill K. [et al.]. Spinal muscular atrophy : Time for newborn screening? Nervenarzt. 2017. Vol. 88, № 12. P. 1358–66.