Background
Limb-girdle muscular dystrophy (LGMD) is an umbrella term given to a diverse group of highly heterogeneous, rare, autosomal neuromuscular disorders with many subtypes categorized by disease genes and inheritance. It is caused by mutations in more than 25 genes that encode numerous components of the myofiber, contractile apparatus, nuclear lamina, sarcolemma, or the cytoplasm. LGMD usually manifests itself in the proximal muscles around the hips and shoulders, causes weakness of the proximal muscles of the shoulder and pelvic girdle.
Currently, LGMD has more than 30 different subtypes, associated with specific gene loci, which manifest themselves by crossed and heterogeneous phenotypes [1, 2]. Conside-ring the genetic and phenotypic heterogeneity of this pathology, LGMD should be considered in almost all patients who complain of primary muscle weakness.
Typical clinical manifestations of limb-girdle muscular dystrophy are gait disturbances (waddling gait, so-called duck-like gait). Waddling gait is an exaggerated alternation of lateral trunk movements with an exaggerated elevation of the hip, suggesting the gait of a duck. Affected individuals may have not only an unusual gait, such as waddling or walking on the balls of their feet, but may also have difficulty running, hyperlordosis in the lumbar spine, winged shoulders, a symptom of flabby arms, muscular hypotonia with hypotrophy, and tendon hyporeflexia that is equal to muscle weakness. LGMD patients may need using their arms to press themselves up from a squatting position because of their weak thigh muscles. As the condition progresses, people with limb-girdle muscular dystrophy may eventually require a wheelchair. Increased levels of creatine phosphokinase (CK) are observed in blood plasma.
There are two major groups of LGMDs. The classification of LGMD into autosomal dominant forms — LGMD-D (or LGMD1 according to the old classification) and autosomal recessive forms — LGMD-R (LGMD2 according to the old classification) is supplemented by a classification based on the involved proteins and major genetic defects [3]. Dominant forms are less common than recessive [4].
The prevalence of LGMD is unknown, but estimates range from one in 14,500 to one in 123,000 and depend on its subtype and geography [5]. It is difficult to determine the prevalence of LGMD because of the variety of its features that is commonly overlapping with other muscle disorders. LGMD is the fourth most common muscular dystrophy after dystrophinopathies, myotonic dystrophy, and facioscapulohumeral dystrophy.
Various forms of LGMD may be inherited as autosomal dominant or recessive traits. Тhere are currently recognized eight subtypes of LGMD1 (A–H), and LGMD2 has 17 subtypes (A–Q). The major amount of LGMD is inherited by autosomal recessive type, and only about 10 % are autosomal dominant forms. Autosomal dominant and autosomal recessive forms of LGMD are equally found in men and women. Most recessive cases of LGMD are caused by calpainopathy, dysferlinopathy, and sarcoglycanopathy [6, 7].
The age at onset, severity, and progression of symptoms of these subtypes may vary greatly from case to case, even among individuals in the same family. The onset age also varies among different mutations and ranges from 1 to 50 years. Signs and symptoms may first appear at any age and generally worsen with time, although in some cases they remain mild. In some patients, LGMD may be asymptomatic. LGMD varies among families and family members with the same mutation. In general, patients with autosomal dominant LGMD have a later onset and slower course than those with autosomal recessive type. The increase in creatine kinase is not as significant in autosomal dominant LGMD as in autosomal recessive forms.
The differential diagnosis of LGMD includes facioscapulohumeral muscular dystrophy, Emery-Dreifuss muscular dystrophy, congenital muscular dystrophy, polymyositis, myotonic, myofibrillar, distal and metabolic myopathy, collagen VI-related disorders and dermatomyositis, endocrine myopathies, storage diseases (in particular, Pompe disease), progressive muscular dystrophy, Leyden-Möbius muscular dystrophy, spinal muscular atrophy, and inflammatory myopathies (dermato-/polymyositis).
The purpose was to provide a detailed presentation of two clinical cases of patients with limb-girdle muscular dystrophy as an example of an integrated approach to the diagnosis of progressive muscular dystrophy using comprehensive clinical, genetic, and neuroimaging analysis.
Materials and methods
The materials and methods of this study were reported previously in detail [8]. Briefly, all study participants were admitted to the Department of Neurology of the Uzhhorod National University, Regional Clinical Center of Neurosurgery and Neurology (Uzhhorod, Ukraine). Clinical history, 12-lead electrocardiogram, blood testing, muscle magnetic resonance imaging (MRI) were obtained for all study participants. Limb-girdle muscular dystrophy was determined according to criteria of the American Academy of Neurology and the American Association of Neuromuscular & Electrodiagnostic Medicine, which were endorsed by the American Academy of Physical Medicine and Rehabilitation, the Child Neurology Society, the Jain Foundation, and the Muscular Dystrophy Association [9], and 229th ENMC international workshop: Limb girdle muscular dystrophies — Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017 [3], and was confirmed by the genetic analysis and neuroimaging. For all identified variants of LGMD, gene mapping was performed on a specific chromosome. For some variants of LGMD, the main types of pathological mutations and protein expression products were identified. Parametric and non-parametric statistical methods were applied.
Results and discussion
The list of genes for LGMD screening is too large, and it is more suitable to apply the targeted next-generation sequencing (NGS) panels, which should include any gene that so far has been associated with the clinical picture of LGMD.
Along with the standard methods of diagnosing muscle pathology that have been in the practice of neurologists for a long time (electromyography, biopsy), the role of MRI is critical. It is used to identify or assess the degree of dystrophic changes in the muscles, which were defined as a “replacement of skeletal muscles with the fat tissues, detected on standard T1-weighted axial images”. In recent decades, MRI has become crucial for the diagnosis of soft tissue diseases. Its value has been convincingly proven in hereditary and inflammatory neuromuscular diseases. In addition to neurological and neurophysiological researches, muscle MRI is a valuable diagnostic tool that allows assessing the degree and, more importantly, narrowing the diagnostic search in the most difficult-to-treat cases, especially muscle diseases [3, 10–12]. MRI of the lower extremity muscles is a reliable non-invasive tool for diagnosing and assessing the progression of neuromuscular diseases, showing specific patterns of muscle damage in many myopathies [14, 15].
MRI helps differentiate specific forms and subtypes of LGMD, assess the severity and distribution of muscle involvement, and, therefore, determine the specific features in many forms of the disease [16, 17]. The change in the hyperintense signal is observed in the affected muscles when performing T1-weighted (T1W) scanning. MRI studies have shown damage to the certain muscle groups in the lower extremities, depending on the type and subtype of LGMD.
/51.jpg)
For example, patients with LGMD2A show marked involvement of the hip adductor, patellar tendon, and medial head, while the function of the tailor’s muscle is preserved, and imaging shows the involvement of the plantaris muscle, vastus medialis, and biceps femoris [18]. Patients with LGMD2B may have a variable MRI pattern, mainly involving the adductor, quadriceps, and tibialis muscles, with relative preservation of the tailor’s and gracilis muscles [19]. The calf muscle is predominantly affected in Miyoshi myopathy, lesions of the gluteal muscles, anterior and posterior thigh muscles are more typical for patients with the LGMD phenotype. Patients with sarcoglycanopathy do not show any significant difference like muscle damage on MRI. The adductors and gluteal muscles are more likely to be affected at the very early stage of the disease that commonly manifested itself by damage to the vastus lateralis muscle. The long adductor muscle may show some intact areas, with complete or relatively intact tibialis posterior and flexor digitorum longus muscles. Relative hypertrophy of the sartorius and the gracilis muscles may be detected [19]. Patients with LGMD2D had more severe MRI changes in anterior vs posterior thigh muscles. Muscle tissue in patients with LGMD2I who have mutation in the fukutin-related protein gene is characterized by a pattern of fatty infiltration and edema on the MRI of the vastus intermedius and vastus medialis muscles [20].
In the article, we present two clinical cases in patients who are receiving treatment at the Regional Clinical Center of Neurosurgery and Neurology in Uzhhorod (hereinafter, Center). Modern diagnostic methods, including muscle MRI and genetic counseling, were used in both patients.
Clinical case 1
Patient 1, 31 years old. She has been under the supervision in the Center for 3 years with complaints about feeling of weakness and tightness in the legs (predominately thighs), gait disturbances, difficulty getting up from a chair and climbing stairs.
From the anamnesis morbi: considers herself ill for the last 12 years, when she began to feel weakness in legs. The weakness increased gradually. With time, the pain in the lower extremities appeared, it became difficult to walk (last 5–7 years), get up from a chair. Difficulty climbing stairs, raising arms (during combing) appeared and increased during the last 3 years. Family history: no known hereditary pathology.
Neurological status. The speech is clear. The face is symmetrical, the tongue is medial. Swallowing is not affected. The pharyngeal reflex is preserved. The mobility of the soft palate during phonation is preserved. Tendon reflexes from the hands are preserved, patellar tendon reflex is absent, Achilles reflexes are with foot clonus. There is hypotonia of the muscles of the lower extremities. Atrophy-hypotrophy was not detected (the measurements of the shin: right and left 33 cm, thighs: right and left 44 cm). Muscle strength in the lower extremities was up to 3.0 points (proximal), 4.0 points distally, in the hands — up to 4.0 points (proximal). Sensitivity is not impaired. Standing on the toes is not impaired while standing on the heels is difficult. A Gowers’ sign is positive. The patient has waddling gait.
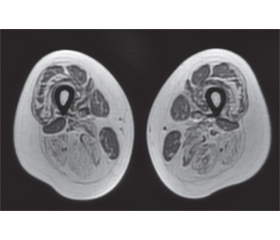
Laboratory and instrumental examination results. CK 929.0, ALT 52.0, AST 47.0. Chest X-ray, echocardiography, spirometry, spirography, cardiological examination are with no detected pathology. Genetic analysis of alpha-galactosidase is negative. MRI of the lumbar spine shows hyperlordosis and a perineural (Tarlov) cyst at S2 level. EMG: no pathology detected in the motor and sensory fibers of the upper and lower extremities; test of neuromuscular transmission— myasthenic block was not detected; EMG signs of generalized, myopathic type, lesions with a predominance in the lower extremities are more proximal. MRI of the pelvic girdle muscles was performed using T1-, T2-weighting and STIR-sequence, following muscles are intact on both sides: m.rectus femoris, m.sartorius, m.gracilis, m.biceps femoris, caput breve; bilateral subtotal adipose tissue replacement of the anterior and posterior thigh muscle fibers was detected, as well as atrophic changes with adipose tissue replacement of the posterior shin muscles bilaterally, stage 1–3 on the Mercuri and Fischer scales (Fig. 1, 2).
The patient was examined by a rheumatologist, as a result, inflammatory myopathies — polymyositis and dermatomyositis — were excluded. During genetic counseling, NGS was performed. A mutation in the calpain-3 (CAPN3) gene was detected in a homozygous state. The CAPN3 gene is associated with the autosomal recessive LGMDR1 (LGMD2A according to the old classification).
Clinical case 2
Patient 2, 23 years old. She visited outpatient clinic for a year with complaints of weakness in the arms, more on the right, inability to raise arms above the head, thinning of the right arm, asymmetry of facial muscles, difficulty walking, difficulty getting out of bed.
From the anamnesis: the patient has been ill since she was about 10 years old when she began to notice the asymmetry of the face on the left and tightening of the lips. In 2015, after childbirth, she began to notice weakness and thinning of the right arm (shoulder/forearm) that has progressed over several months, the weakness in the left arm appeared. Over the past year, the weakness in the upper extremities increased significantly, especially on the right, the patient noted difficulty to get out of bed, inability to raise her arms above the chest. Family history: without hereditary pathology.
Neurological status. The speech is clear. The face is asymmetrical, hypotrophy of facial muscles on the left, patient cannot pucker her lips. Swallowing is not disturbed. The pharyngeal reflex is preserved. Tendon reflexes from the hands are torpid, D = S, patellar and Achilles are restored. Hypotrophy of the right shoulder girdle muscles, winged scapula. Hypotonia of the muscles of the upper extremities, more on the right. Muscle strength in the extremities: in the hands proximally — to 2.0 points, distally — to 3.0 points; in the extensors of the right foot — up to 3 points. Standing on the toes is preserved, on the heels is not possible. A Gowers’ sign is negative.
/52.jpg)
Laboratory and instrumental examination results. СK 834.0, ALT — 32.3, AST — 30.4. Chest X-ray, echocardiography, spirometry and spirography, cardiological examination — without pathology. Genetic analysis of alpha-galactosidase deficiency is negative. Consulted by a cardiologist — sinus arrhythmia. ENMG: signs of the reorganization of ABM on myopathic type mainly in the upper extremities, more proximal departments, with a slight asymmetry more on the right. Spontaneous activity is absent (fibrillation and fasciculation potentials were not obtained). MRI of the pelvic, shoulder, and girdle muscles was performed using T1-, T2-weighting, and STIR-sequence: atrophic changes of the upper shoulder girdle muscles, on the right: m.trapezius, semispinalis capitis et cervicis, m.serratus anterior, supraspinalis capitis et cervicis were found; atrophic changes due to the replacement of adipose tissue fibers of the thigh muscles, mainly the posterior and medial groups; the anterior group of thigh muscles was almost intact. Atrophic changes in the tibialis anterior muscle are noted; stage 2–3 on the Mercuri and Fischer scales (Fig. 3, 4).
Patient underwent genetic counseling, the NGS panel was used. The diagnostic search included the gene of facioscapulohumeral muscular dystrophy, taking into account the asymmetry of the lesion. The carrier of the SGCG gene in the heterozygous state was detected. The SGCG gene is associated with the autosomal recessive LGMDR5 (LGMD2C according to the old classification) — γ-sarcoglycanopathy.
Conclusions
Thus, given the clinical heterogeneity of progressive muscular dystrophies, the diagnosis should be based on both additional methods and, of course, the clinical picture of the disease. Taking into account our experience, patients with myopathy syndrome require a comprehensive clinical and diagnostic approach, including the involvement of related specialists. It is necessary to conduct a complete clinical and biochemical examination, including determination of the level of CK, ALT, AST. The results of laboratory tests need to be carefully compared with the results of clinical and instrumental examination, in particular with the data of ENMG and MRI of muscles. Clinical use of muscle MRI and ENMG helps in the early diagnosis of progressive forms of muscular dystrophies that will allow for timely treatment and rehabilitation measures in the future.
Conflicts of interests. Authors declare the absence of any conflicts of interests and their own financial interest that might be construed to influence the results or interpretation of their manuscript.
Additional information
Role of the funding source — none
Disclosures — none
Author contributions: H.V. Palahuta — study concept and design, data acquisition, statistical analysis, interpretation of the data, literature overview, critical revision of the manuscript for important intellectual content; O.Ye. Fartushna — literature overview, statistical analysis, interpretation of data, article concept, and design, drafting the article, critical revision of the manuscript for important intellectual content.
Список литературы
1. Nigro V., Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 2014. Vol. 33. P. 1-12.
2. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С., Евтушенко И.С. Нейромышечные заболевания у детей: монография. Донецк: Изд-во «Ноулидж» (донецкое отделение), 2014. 218 с.
3. Straub V., Murphy A., Udd B. LGMD workshop study group 229th ENMC international workshop: Limb girdle muscular dystrophies — Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March, 2017. Neuromuscul. Disord. 2018. Vol. 28. P. 702-710.
4. Mahmood O.A., Jiang X.M. Limb-girdle muscular dystrophies: where next after six decades from the first proposal (Review). Mol. Med. Rep. 2014. Vol. 9. P. 1515-1532.
5. Liewluck T., Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle & Nerve. 2018. Vol. 58, № 2. P. 167-177.
6. Magri F., Nigro V., Angelini C. et al. The Italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle & Nerve [Internet]. 2017. Vol. 55. P. 55-68.
7. Bushby K. Diagnosis and management of the limb girdle muscular dystrophies. Pract. Neurol. 2009. Vol. 9, № 6. P. 314-323.
8. Palahuta H.V., Smolanka V.I., Dutka I.Yu. The diagnostic role of magnetic resonance imaging of muscles in neuromuscular diseases: scientific review and personal observation. International Neurological Journal. 2019. Vol. 1, № 103. P. 10-14.
9. Narayanaswami P., Weiss M., Selcen D. et al. Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2014. Vol. 83, № 16. P. 1453-1463.
10. Влодавец Д.В., Казаков Д.О. Диагностические возможности магнитно-резонансной томографии мышц при нервно-мышечных заболеваниях. Неврологический журнал. 2014. № 3. С. 4-12.
11. Carlier P.G., Marty B., Scheidegger O., Loureiro de Sousa P., Baudin P., Snezhko E., Vlodavets D. Skeletal muscle quantitative nuclear magnetic resonance imaging and spectroscopy as an outcome measure for clinical trials (part I). Neuromuscular Diseases. 2016. Vol. 4. P. 10-20.
12. Carlier P.G., Marty B., Scheidegger O., Loureiro de Sousa P., Baudin P., Snezhko E., Vlodavets D. Skeletal muscle quantitative nuclear magnetic resonance imaging and spectroscopy as an outcome measure for clinical trials (part II). Neuromuscular Diseases. 2017. Vol. 7, № 1. P. 11-29.
13. Tasca G., Monforte M., Iannaccone E. et al. Muscle MRI in female carriers of dystrophinopathy. European Journal of Neurology. 2012. Vol. 19, № 9. P. 1256-1260.
14. Lovitt S., Moore S.L., Franklin A., Marden F.A. The use of MRI in the evaluation of myopathy. Clinical Neurophysiology. 2006. Vol. 117. P. 486-495.
15. Мercuri E., Clements E. et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann. Neurol. 2010. Vol. 67, № 2. P. 201-208.
16. Wattjes M.P., Kley R.A., Fischer D. Neuromuscular imaging in inherited muscle diseases. Eur. Radiol. 2010. Vol. 20. P. 2447-2460.
17. Ten Dam L., Van der Kooi A.J., Van Wattingen M. et al. Reliability and accuracy of skeletal muscle imaging in limb girdle muscular dystrophies. Neurology. 2012. Vol. 79. P. 1716-1723.
18. Richard I., Hogrel J.Y., Stockholm D., Payan C.A., Fougerousse F., Calpainopathy Study Group, et al. Natural history of LGMD2A for delineating outcome measures in clinical trials. Ann. Clin. Transl. Neurol. 2016. Vol. 3, № 4. P. 248-265.
19. Jin S., Du J., Wang Z., Zhang W., Lv H., Meng L. et al. Heterogeneous characteristics of MRI changes of thigh muscles in patients with dysferlinopathy. Muscle & Nerve. 2016. Vol. 54, № 6. P. 1072-1079.
20. Tasca G., Monforte M., Díaz-Manera J. et al. MRI in sarcoglycanopathies: a large international cohort study. J. Neurol. Neurosurg. Psychiatry. 2018. Vol. 89, № 1. P. 72-77.