Вступ
IgG4-залежне захворювання (IgG4-ЗЗ) — це імуноопосередкований стан, асоційований з утворенням фіброзно-запальних мас, що можуть виникати практично в будь-якому органі та імітувати злоякісні, інфекційні та запальні захворювання [1]. Уніфікована номенклатура IgG4-ЗЗ була прийнята лише у 2012 році [2]. Цього ж року були запропоновані діагностичні критерії IgG4-ЗЗ [3]. У 2015 році були розроблені міжнародні консенсусні рекомендації щодо ведення хворих на IgG4-ЗЗ [4]. У 2019 році на щорічному з’їзді Американського коледжу ревматологів (ACR) та Європейської антиревматичної ліги (EULAR) були затверджені класифікаційні критерії IgG4-ЗЗ [1].
IgG4-ЗЗ вважається рідкісним захворюванням, але його реальна епідеміологія досі не визначена. Доступні епідеміологічні дані базуються переважно на японських популяційних дослідженнях: за даними Uchida et al. (2012), річна поширеність IgG4-ЗЗ становить 0,28–1,08 випадку на 100 000 населення [5]. IgG4-ЗЗ зазвичай уражує осіб середнього та старшого віку з дебютом переважно у віці 50–70 років, хоча описані рідкісні випадки в дітей.
IgG4 — найменш поширений підклас IgG, що становить менше 5 % від загального IgG (норми його сироваткового рівня коливаються від 0,05 до 1,4 г/л). Підвищення рівня IgG4 у сироватці крові донедавна відігравало основну роль у встановленні діагнозу IgG4-ЗЗ, але дані останніх років свідчать про його низьку діагностичну цінність. Так, у 30 % хворих із IgG4-ЗЗ визначаються нормальні рівні IgG4 у сироватці крові. З іншого боку, підвищений рівень IgG4 може спостерігатися при багатьох інших захворюваннях: синдромі Шегрена, системному червоному вовчаку, ревматоїдному артриті, раку, хворобі Кастлемана, алергічних захворюваннях, еозинофільному гранулематозі з поліангіїтом, саркоїдозі, а також у здоровій популяції (2 %) [6].
Патоморфологія, класифікація та клінічні прояви IgG4-ЗЗ
Ключовими морфологічними ознаками IgG4-ЗЗ є: 1) щільний лімфоплазмоцитарний інфільтрат з високим вмістом IgG4+-плазматичних клітин; 2) спіралеподібний фіброз; 3) флебіт з облітерацією просвіту судин. Інші гістопатологічні зміни представлені флебітом без облітерації просвіту судин і з підвищеним вмістом еозинофілів. Важливе значення має імуногістохімічне дослідження з кількісним визначенням IgG4+-плазматичних клітин [6].
Досі немає загальноприйнятої класифікації IgG4-ЗЗ. Перелік нозологічних одиниць щорічно переглядається та доповнюється (табл. 1).
Початок IgG4-ЗЗ зазвичай є підгострим. У хворих часто відсутня будь-яка симптоматика, і захворювання діагностується випадково під час обстеження з приводу інших причин [7]. Можлива також спонтанна ремісія з багаторічною відсутністю активності захворювання [8]. Важливою ознакою захворювання є позитивна клінічна відповідь на терапію ex juvantibus глюкокортикоїдами (ГК) [6].
Хвороба була зареєстрована практично в кожній системі органів [8]. Приблизно у 40 % хворих захворювання дебютує з ураження одного органа в формі об’ємного утворення, що викликає специфічні прояви залежно від локалізації. Симптоми варіюють від набряку уражених органів (слинних і слізних залоз, лімфатичних вузлів) до обструкції (проток підшлункової залози, сечоводів), дисфункції органів (гіпофізарна недостатність внаслідок гіпофізиту, ниркова недостатність) і навіть невідкладних станів (гострий аортальний синдром, пахіменінгіт, панкреатит). У невеликої кількості хворих спостерігаються конституційні симптоми, зокрема лихоманка та схуднення [9].
Діагностика IgG4-ЗЗ
Для діагностики IgG4-ЗЗ використовують діагностичні критерії H. Umehara et al. (2012), що базуються на клінічних, імунологічних і гістопатологічних ознаках (табл. 2).
У 2019 році на щорічному з’їзді EULAR/ACR у Чикаго були затверджені класифікаційні критерії IgG4-ЗЗ [1]. Першим кроком у встановленні діагнозу IgG4-ЗЗ є ідентифікація клінічного/рентгенологічного ураження типового органа (підшлункова залоза, жовчні протоки, орбіти, слізні залози, великі слинні залози, заочеревинний простір, нирки, аорта, м’які оболонки мозку, щитоподібна залоза). У пацієнтів без залучення принаймні одного з цих органів виключається ймовірність IgG4-ЗЗ. Наступним кроком є перевірка відсутності у пацієнта критеріїв виключення (табл. 3). Завершальним етапом є виявлення достатньої кількості класифікаційних ознак для остаточного підтвердження IgG4-ЗЗ (табл. 4).
IgG4-залежне захворювання нирок (IgG4-ЗЗН)
Ураження нирок спостерігається у близько 15 % хворих на IgG4-ЗЗ [10]. Хворіють переважно чоловіки (73–87 %), середній вік — 65 років [11]. У пацієнтів часто наявні супутні позаниркові прояви (сіалоаденіт, панкреатит, лімфаденопатія). Характерним є підвищення рівня загального сироваткового IgG, IgG4, IgE, креатиніну [10]. Зміни маркерів запального процесу, таких як ШОЕ і С-реактивний білок, не є характерними для цього захворювання і не корелюють з його активністю. Це важливо враховувати під час проведення диференціальної діагностики з АНЦА-асоційованими васкулітами і мультицентричною хворобою Кастлемана [12]. Підвищений рівень IgG4 у сироватці крові спостерігається практично у всіх хворих з IgG4-ЗЗН, тоді як у близько 30 % хворих з IgG4-ЗЗ без ураження нирок концентрація IgG4 залишається нормальною [11]. Рівень IgG4 різко знижується після успішної терапії ГК, але повторно підвищується приблизно у половини хворих на фоні підтримуючої терапії ГК без очевидних рецидивів захворювання [13].
Основним інструментальним методом обстеження при IgG4-ЗЗН є комп’ютерна томографія (КТ) з контрастуванням, що використовується для оцінки поширеності процесу та контролю за перебігом захворювання. Корисною є також позитронно-емісійна томографія (ПЕТ) з 18-фтордезоксиглюкозою, що дозволяє виявити активні запальні ураження, оцінити ступінь поширеності захворювання та реакцію на лікування, а також визначити точне місце для біопсії [6].
Розрізняють три основні форми IgG4-ЗЗН [10]:
1) IgG4-залежний тубулоінтерстиціальний нефрит (ТІН);
2) IgG4-залежний мембранозний гломерулонефрит (МГН);
3) вторинний гідронефроз внаслідок стиснення сечоводів при IgG4-залежному ретроперитонеальному фіброзі.
ТІН є найпоширенішою формою ураження нирок при IgG4-ЗЗ. У більшості випадків ТІН діагностують на фоні вже підтверджених екстраренальних проявів IgG4-ЗЗ (у 39 % хворих наявний автоімунний панкреатит 1-го типу). Клінічно ураження нирок може проявлятись порушенням функції нирок з легкою/помірною протеїнурією, в рідких випадках можлива гематурія [11]. На відміну від медикаментозно-індукованого ТІН IgG4-залежний ТІН переважно не супроводжується лейкоцитурією або появою лейкоцитарних циліндрів у сечі [13]. Нефротичний синдром не є характерним для IgG4-залежного ТІН, за його наявності слід запідозрити супутній МГН. У 60 % хворих із IgG4-залежним ТІН наявна гіпокомплементемія (зниження СН50, С3, С4), що не є характерним для IgG4-ЗЗ без ураження нирок. Повторне зниження рівня комплементу у хворих, у яких була гіпокомплементемія до початку терапії, асоціюється з можливим рецидивом IgG4-ЗЗН. У 30 % пацієнтів можливе виявлення АНА в низьких титрах без специфічного спектра антитіл. Крім того, у 40 % хворих наявні периферична еозинофілія та підвищення сироваткового IgE [11].
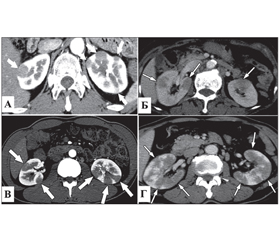
Найпоширенішими проявами IgG4-залежного ТІН на КТ є множинні двосторонні вогнища низької щільності, переважно у кірковому шарі нирок, що спостерігаються у близько 40 % пацієнтів [10]. Розрізняють 4 основні КТ-патерни вогнищ при IgG4-залежному ТІН (рис. 1):
1) периферичні вузлики < 1 см у кірковому шарі (рис. 1А);
2) чіткі або малочіткі округлі вогнища (рис. 1Б);
3) клиноподібні вогнища (рис. 1В);
4) дифузні неоднорідні вогнища (рис. 1Г).
Іншим можливим проявом IgG4-залежного ТІН є потовщення ниркової миски (рис. 2А), що потребує диференціальної діагностики з пієлонефритом, лімфомою, метастазами пухлин, емболічними ураженнями. У 20 % хворих спостерігається дифузне збільшення обох нирок (рис. 2В). Можливе також виявлення об’ємного пухлиноподібного утворення, що виходить за межі нирки та нагадує нирково-клітинну карциному (рис. 2Б) [10].
/91.jpg)
Для IgG4-залежного ТІН характерні класичні гістологічні ознаки IgG4-ЗЗ, хоча облітеруючий флебіт спостерігається порівняно рідко. Типовими ознаками є зональний ТІН з інфільтрацією IgG4-позитивними клітинами, спіралеподібним фіброзом і атрофією канальців. Методом імунофлуоресцентної та електронної мікроскопії (ЕМ) виявляють відкладання депозитів імунних комплексів (ІК) у базальній мембрані (БМ) канальців (наведені переважно IgG, C3, C1q). Імуногістохімічне дослідження дозволяє виявити типові для IgG4-ЗЗ зміни: співвідношення IgG4/IgG+ плазмоцитів > 40 %; кількість IgG4+-плазмоцитів > 10 в полі зору (п/з) [10].
Існує 2 види діагностичних критеріїв IgG-залежного ТІН, кожен з яких базується на гістологічних, візуалізаційних, серологічних і клінічних ознаках (табл. 5) [15].
Вторинний МГН є єдиним варіантом ураження клубочків нирок при IgG-ЗЗ і спостерігається приблизно у 7 % хворих з IgG4-ЗЗН [10]. У 50 % пацієнтів наявний супутній ТІН (на відміну від ідіопатичного МГН). Вторинний МГН часто супроводжується нефротичним синдромом. При гістологічному дослідженні рідко спостерігаються лімфоплазмоцитарна інфільтрація та спіралеподібний фіброз. При імунофлуоресцентному дослідженні та ЕМ виявляють субепітеліальні депозити мембранозного патерну (представлені переважно IgG4), при цьому відсутні депозити антитіл до рецептора секреторної фосфоліпази А2 (PLA2R), що важливо враховувати під час диференціальної діагностики первинного й IgG4-залежного МГН (табл. 6) [10].
/92.jpg)
Згідно з алгоритмом діагностики японських дослідників, наявність пошкодження нирок (зміни в загальному аналізі сечі, рентгенологічні прояви, зниження функції нирок) у поєднанні з підвищенням рівня загального сироваткового IgG/IgE або гіпокомплементемією є першим етапом, на якому слід запідозрити IgG4-ЗЗН (рис. 3). Після виключення інших захворювань (системний червоний вовчак, системні васкуліти) необхідно підтвердити підвищення рівня IgG4 у сироватці крові. Наступним етапом є оцінка характерних рентгенологічних і гістологічних проявів. У пацієнтів з неможливістю проведення біопсії нирки діагноз IgG4-ЗЗН базується на основі типових рентгенологічних проявів за умови гістологічно підтвердженого IgG4-залежного ураження інших органів [11].
/93.jpg)
Ретроперитонеальний фіброз (РПФ) — хронічне запальне захворювання з вираженим фіброзом тканин заочеревинного простору. РПФ здебільшого виникає у чоловіків середнього віку та асоціюється з палінням. Клінічно у хворих з двобічним масивним розростанням щільної волокнистої сполучної тканини в ретроперитонеальній клітковині виникає двостороння обструкція сечоводів з розвитком больового синдрому й анурії (рис. 4А). IgG4-залежний аортит зазвичай має безсимптомний перебіг і може дебютувати розривом аневризми аорти. Характерною ознакою IgG4-залежного аортиту, за даними контрастної КТ, є кругове потовщення артеріальної стінки, що обумовлено запаленням і склерозом адвентиції (рис. 4Б), та накопичення 18-фтордезоксиглюкози за даними ПЕТ [6].
Особливості лікування IgG4-ЗЗН
Тактика ведення хворих з IgG4-ЗЗН загалом відображає принципи лікування IgG4-ЗЗ [4, 18–22]. Навіть субклінічне ураження нирок може призвести до тяжких необоротних ушкоджень протягом кількох місяців і тому потребує негайного лікування. У деяких випадках виникає потреба в терміновому хірургічному втручанні, зокрема при обструкції сечоводів [6].
ГК є препаратами першої лінії для індукції ремісії у всіх хворих з IgG4-ЗЗН за відсутності протипоказань. Здебільшого для стартової терапії використовують преднізолон в дозі 0,6 мг/кг на добу. Відповідь на терапію ГК зазвичай спостерігається протягом кількох тижнів: знижується рівень сироваткового креатиніну, підвищується концентрація комплементу та нормалізується рівень IgG4. Цікаво, що відповідь на терапію ГК не корелює з гістологічним патерном, оскільки, за даними клініки Мейо, навіть у пацієнтів з вираженим інтерстиційним фіброзом була хороша відповідь на лікування ГК [21]. У хворих з початковими проявами IgG4-залежного ТІН після успішного лікування ГК рентгенологічні зміни часто повністю зникають без рубцювання чи атрофії [12]. Однак у значної кількості пацієнтів навіть після успішного лікування може виникати вогнищева або дифузна атрофія кіркового шару нирок (рис. 5). Якщо на момент початку лікування швидкість клубочкової фільтрації становить < 60 мл/хв, функція нирок відновлюється лише частково (переважно в перший місяць, а потім утримується на одному рівні). Описані випадки розвитку термінальної стадії ниркової недостатності, що потребувало замісної ниркової терапії [12]. У близько 30 % пацієнтів після припинення лікування ГК виникають рецидиви, тому японські дослідники рекомендують постійну підтримуючу терапію ГК [12]. Предикторами рецидивів є повторна поява гіпокомплементемії [12], протеїнурії, підвищених рівнів IgG4, IgE, циркулюючих еозинофілів [20–21].
/94.jpg)
Експертні думки залишаються невизначеними щодо питання, чи слід застосовувати імуносупресивні засоби в індукційній і підтримуючій терапії, окрім ГК. Згідно з консенсусними рекомендаціями, використання імуносупресорів доцільне у випадках, коли доза ГК не може бути знижена через постійно високу активність захворювання. Серед цитостатиків для лікування IgG4-ЗЗ використовують азатіоприн, мікофенолату мофетил, метотрексат, 6-меркаптопурин, такролімус і циклофосфамід. Проте ефективність цих препаратів не була оцінена в проспективних дослідженнях [9].
У хворих з рецидивним перебігом IgG4-ЗЗН або при стероїдорезистентності можливе застосування ритуксимабу (інгібітор СD20+ В-лімфоцитів; RTX) [20–23]. За даними M. Ebbo et al. (2017), клінічна відповідь була отримана у 93,5 % пацієнтів з IgG4-ЗЗ через місяць після початку терапії RTX. Наприкінці дослідження відміна ГК була досягнута у 51,5 % пацієнтів; середня підтримуюча доза ГК становила 9,6 ± 9,3 мг/добу.
Проте протягом середнього періоду спостереження (24,8 ± 21,0 міс.) у 41,9 % пацієнтів відбувся рецидив захворювання через 19 ± 11 міс. після припинення терапії RTX. Натомість підтримуюча терапія RTX вірогідно сприяла тривалішому безрецидивному періоду (р = 0,02) [24]. І хоча результати останніх досліджень є перспективними, досі відсутній консенсус щодо дозування та частоти застосування RTX при IgG4-ЗЗН.
Висновки
Отже, ураження нирок є одним із найчастіших проявів IgG4-ЗЗ. Клінічна картина на початку захворювання часто невиражена, тому важлива роль у встановленні діагнозу належить інструментальним методам дослідження. Зокрема, типовими КТ-ознаками IgG4-ЗЗН є множинні двосторонні вогнища низької щільності у кірковому шарі, потовщення ниркової миски, дифузне збільшення обох нирок, а також виявлення об’ємного пухлиноподібного утворення. Ураження нирок при IgG4-ЗЗ часто супроводжується гіпокомплементемією, підвищенням сироваткового IgG4, IgE, периферичною еозинофілією. Для IgG4-ЗЗН характерна хороша відповідь на терапію ГК, проте навіть незначна затримка в лікуванні може призвести до виникнення тяжких необоротних ушкоджень ниркової тканини.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів і власної фінансової зацікавленості при підготовці цієї статті.
Отримано/Received 05.03.2021
Рецензовано/Revised 19.03.2021
Прийнято до друку/Accepted 25.03.2021
Список литературы
1. Wallace Z.S., Naden R.P., Chari S. et al. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Annals of the Rheumatic Diseases. 2020. № 79(1). P. 77-87. doi: 10.1136/annrheumdis-2019-216561.
2. Stone J.H., Khosroshahi A., Deshpande V. et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis and rheumatism. 2012. № 64(10). P. 3061-3067. doi: 10.1002/art.34593.
3. Umehara H., Okazaki K., Masaki Y. et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD). Mod. Rheumatol. 2012. № 22. P. 21-30. doi: 10.1007/s10165-011-0571-z.
4. Khosroshahi A.M., Wallace Z.S., Crowe J.L. et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease.Arthritis Rheumatol. 2015. № 67(7). P. 1688-1699. doi: 10.1002/art.39132.
5. Uchida K., Masamune A., Shimosegawa T., Okazaki K. Prevalence of IgG4-Related Disease in Japan Based on Nationwide Survey in 2009. International journal of rheumatology. 2012. 358371. doi: 10.1155/2012/358371.
6. Iaremenko O.B., Koliadenko D.I., Petelytska L.B. IgG4-related disease: current state of the problem and a description of the clinical case. Ukrainskyi revmatolohichnyi zhurnal. 2019. № 75(1). P. 10-19. (In Ukrainian).
7. Sebastian A., Sebastian M., Misterska Skóra M. et al. The variety of clinical presentations in IgG4-related disease in Rheumatology. Rheumatol. Int. 2018. № 38(2). P. 303-309. doi: 10.1007/s00296-017-3807-1.
8. Sedhom R., Sedhom D., Strair R. IgG4-related disease: A mini-review. J. Rare Dis. Res. Treat. 2017. № 2(2). P. 18-23. doi: 10.29245/2572-9411/2017/2.1089.
9. Weindorf S.C., Frederiksen J.K. IgG4-Related Disease: A Reminder for Practicing Pathologists. Arch Pathol Lab Med. 2017. № 141(11). P. 1476-1483. doi: 10.5858/arpa.2017-0257-RA.
10. Cortazar F.B., Stone J.H. IgG4-related disease and the kidney. Nat. Rev. Nephrol. 2015. № 11(10). P. 599-609. doi: 10.1038/nrneph.2015.95.
11. Saeki T., Kawano M. IgG4-related kidney disease. Kidney Int. 2014. № 85(2). P. 251-217. doi: 10.1038/ki.2013.393.
12. Kawano M., Yamada K. IgG4-Related Kidney Disease and IgG4-Related Retroperitoneal Fibrosis. Semin Liver Dis. 2016. № 36(3). P. 283-90. doi: 10.1055/s-0036-1584316.
13. Kawano M., Saeki T. IgG4-related kidney disease — an update. Curr. Opin Nephrol. Hypertens. 2015. № 24(2). P. 193-201. doi: 10.1097/MNH.0000000000000102.
14. Oh J.W., Rha S.E., Choi M.H., Oh S.N., Youn S.Y., Choi J.I. Immunoglobulin G4-related Disease of the Genitourinary System: Spectrum of Imaging Findings and Clinical-Pathologic Features. Radiographics : a Review Publication of the Radiological Society of North America, Inc. 2020. № 40(5). P. 1265-1283. doi: 10.1148/rg.2020200043.
15. Salvadori M., Tsalouchos A. Immunoglobulin G4-related kidney diseases: An updated review. World J. Nephrol. 2018. Vol. 6. № 7(1). P. 29-40. doi: 10.5527/wjn.v7.i1.29.
16. Zhang N.N., Wang Y.Y., Kong L.X., Zou W.Z., Dong B. IgG4-related kidney disease (IgG4-RKD) with membranous nephropathy as its initial manifestation: report of one case and literature review. BMC Nephrol. 2019. № 20(1). P. 263. doi: 10.1186/s12882-019-1419-6.
17. Fujimori N., Ito T., Igarashi H. et al. Retroperitoneal fibrosis associated with immunoglobulin G4-related disease. World J. Gastroenterol. 2013. № 19(1). P. 35-41. doi: 10.3748/wjg.v19.i1.35.
18. Eroglu E., Sipahioglu M.H., Senel S. et al. Successful treatment of tubulointerstitial nephritis in immunoglobulin G4-related disease with rituximab: A case report. World J. Clin. Cases. 2019. № 7(16). P. 2309-2315. doi: 10.12998/wjcc.v7.i16.2309.
19. Nada R., Ramachandran R., Kumar A. et al. IgG4-related tubulointerstitial nephritis: A prospective analysis. Int. J. Rheum. Dis. 2016. № 19. P. 721-729. doi: 10.1111/1756-185X.12675.
20. Pradhan D., Pattnaik N., Silowash R., Mohanty S.K. IgG4-related kidney disease — A review. Pathol. Res. Pract. 2015. № 211(10). P. 707-11. doi: 10.1016/j.prp.2015.03.004.
21. Boffa J.-J. Esteve E., Buob D. Renal involvement in IgG4-related disease. La Presse Médicale. 2020. № 49. 104017. doi: 10.1016/j.lpm.2020.104017.
22. Mann S., Seidman M.A., Barbour S.J., Levin A., Carruthers M., Chen L.Y. Recognizing IgG4-related tubulointerstitial nephritis. Can. J. Kidney Health Dis. 2016. № 17(3). P. 34. doi: 10.1186/s40697-016-0126-5.
23. Teng F., Lu H., Zheng K. et al. Urinary System Manifestation of IgG4-Related Disease: Clinical, Laboratory, Radiological, and Pathological Spectra of a Chinese Single-Centre Study. J. Immunol. Res. 2020. № 2020. 5851842. doi: 10.1155/2020/5851842.
24. Ebbo M., Grados A., Samson M. et al. Long-term efficacy and safety of rituximab in IgG4-related disease: Data from a French nationwide study of thirty-three patients. PLoS One. 2017. № 12(9). e0183844. doi: 10.1371/journal.pone.0183844.

/88.jpg)
/89.jpg)
/90.jpg)
/91.jpg)
/92.jpg)
/93.jpg)
/94.jpg)