Статья опубликована на с. 129-132
Ребенок С., мальчик, 1 мес. 8 дней, поступил в отделение патологии новорожденных ОДКБ № 1 с жалобами на сухость и шелушение кожных покровов, покраснение глаз, вялость, снижение аппетита, отсутствие прибавки массы тела.
Из анамнеза известно, что ребенок родился от II нормально протекавшей беременности, II родов. В течение беременности мать регулярно посещала женскую консультацию, достаточно полно обследована, патологии не выявлено. Роды в срок 38 недель, в родах — преэклампсия, родоразрешение путем кесарева сечения. Оценка по шкале Апгар 7–8 баллов. Масса тела 3150 г, длина тела 49 см, окружность головы 32 см, окружность груди 30 см.
С момента рождения отмечены изменения кожных покровов: резкая гиперемия кожи, к концу первых суток появились единичные пузыри, после вскрытия которых образовались эрозии. В дальнейшем кожа стала сухой, появилось крупнопластинчатое шелушение. У ребенка диагностирована внутриутробная инфекция, эксфолиативный дерматит Риттера. Терапия включала цефосульбин, цефикс, бифидоформ, инфузионная терапия — 5% глюкозу, физиологический раствор; ребенок получал фенистил, витамин А, витамины группы В, -увлажняющие кремы. Существенной положительной динамики в состоянии ребенка не отмечено, на 26-й день жизни мальчик по настоянию матери выписан домой.
В течение последующих 12 дней ребенок находился под наблюдением врачей районной детской поликлиники: педиатра, невролога, лор-врача, офтальмолога. Отмечено нарастание тяжести состояния, заключавшееся в усилении вялости, дальнейшей потере массы тела, усилении гиперемии и сухости кожных покровов, появлении гнойного отделяемого из глаз и наружных слуховых проходов. Дифференциальная диагностика проводилась между инфекционным заболеванием и врожденной патологией. Терапия включала антибиотики, глазные, ушные капли с антибиотиками, мази, обладающие увлажняющим действием. Лечение было неэффективным. Ребенок направлен на обследование и лечение в Харьковскую ОДКБ № 1.
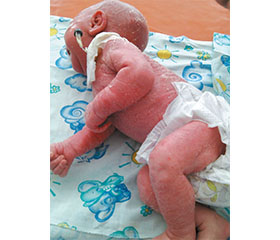
При поступлении состояние ребенка оценено как тяжелое за счет неврологических расстройств, интоксикации в связи с наличием воспалительных очагов — на коже, в глазах, ушах. Ребенок вял, адинамичен, периодически — резко беспокоен. Грудь сосет вяло. Подкожная клетчатка истончена, тургор и эластичность снижены. Дефицит массы тела 26 %. Отмечается эритема всей поверхности кожи, больше выраженная в области живота и ягодиц. Имеют место ихтиозоподобные изменения кожи: сухость, наличие тонких чешуек, выражено шелушение, имеется утолщение кожи на подбородке и вокруг рта, фолликулярный кератоз на конечностях. Особенности волосяного покрова: алопеция, редкие волосы на бровях, ресницы, на теле волосы отсутствуют. Ногти на руках гипопластичны. Рефлексы периода новорожденности снижены. Мышечная гипотония.
Выражена гиперваскуляризация роговицы глаз, дефект роговицы в правом глазу, имеется скудное серозно-гнойное отделяемое из глазных щелей с обеих сторон. В слуховых проходах также серозно-гнойное отделяемое.
На громкий хлопок в ладоши в области обоих ушей ребенок не реагирует. В легких перкуторно ясный легочный звук, при аускультации пуэрильное дыхание. Частота дыхания 48 дыхательных движений в минуту. Границы относительной сердечной тупости в пределах возрастной нормы. Тоны сердца чистые, ясные, ритмичные, 146 уд/мин. Живот мягкий, безболезненный. Печень пальпируется на 2,5 см ниже края реберной дуги, селезенка не пальпируется, наружные половые органы развиты по мужскому типу, стул 1 раз в сутки, кашицеобразный, желтый, без патологических примесей, мочеиспускание свободное, до 7 раз в сутки.
Учитывая время манифестации признаков заболевания — от момента рождения, характер изменений кожи, развитие в динамике заболевания конъюнктивита, отита, был поставлен предварительный диагноз: эксфолиативный дерматит Риттера, гнойный конъюнктивит с перфорацией роговицы справа, двусторонний отит, постнатальная гипотрофия II ст., сепсис под вопросом.
В клинике проведены: консультация офтальмолога, дерматолога, генетика, лор-врача. Произведены: клинический анализ крови, мочи, копрограмма, бактериологические исследования отделяемого из глаз, ушей, из зева, носа, посев крови на стерильность, посев кожи с пораженных участков на флору и чувствительность к антибиотикам.
Заключение офтальмолога: гиперваскуляризация, перфорация роговицы справа с выпадением радужки и десцеметоцеле роговицы слева. Показано проведение в ургентном порядке блефарорафии справа.
Через 45 мин после поступления ребенка в стационар произведено сшивание век правого глаза, назначенная терапия — глазные капли в левый глаз — выполняется.
Заключение лор-врача: двусторонний гнойный наружный отит, назначено лечение.
Результаты лабораторно-инструментальных исследований
В клиническом анализе крови при поступлении: анемия, гемоглобин 74 г/л, лейкоцитоз 19,7 • 109/л, нейтрофилез 79 %, сдвиг формулы влево до миелоцитов. Через 3 дня лейкоцитоз, 37,6 • 109/л, сохраняется нейтрофилез со сдвигом влево. Биохимический анализ крови: общий белок, глюкоза, мочевина, остаточный азот, креатинин, билирубин, АЛТ, АСТ — в пределах возрастной нормы. Клинический анализ мочи, копрограмма без патологии. Результаты бактериологического исследования: отделяемое из глазных щелей — выделен St.saprophyticus, отделяемое из слуховых проходов — E.сoli, посев с кожи — St.saprophyticus, E.facium, Kl.pneumonium, посев из зева и носа — St.aureus, St.saprophyticus, кровь на стерильность — посев роста не дал. Иммунограмма — в пределах возрастной нормы. R-грамма органов грудной клетки: очагово-инфильтративные изменения не выявлены. УЗИ: головной мозг — перивентрикулярное уплотнение. Сердце — ФОО 2,6 мм. Селезенка — очаговые уплотнения. Печень, почки — без особенностей.
Ребенок получал лечение: антибиотики — фортум, амицил, мепенам, флоксиум, цефосульбин. Противогрибковый препарат — флуконазол. Инфузионная терапия — глюкозосолевыми растворами. С иммунозаместительной целью — биовен моно 2 курса. С заместительной целью — отмытые эритроциты 1 раз. Глазные капли офтавикс, тиотриазолин, сенсевит, мазь флоксал. Ушные капли атофэ. Местно на кожу увлажняющие средства — ойлатум крем, локобейз крем, лескинар, ксемоз. Купание с применением липикара флюида.
/130.jpg)
На протяжении двух недель в больнице отмечена некоторая положительная динамика: уменьшилась выраженность эритемы кожных покровов, исчезли проявления отита, ребенок стал активнее сосать грудь, начал прибавлять в весе. Изменения со стороны глаз, однако, оставались серьезными: веки сшиты, произошла перфорация роговицы во втором глазу, эпителизация роговицы не наступает. Ребенок по-прежнему не реагирует на громкий хлопок рядом с ушами. Сохраняется сухость и шелушение кожи, выражены изменения волосяного покрова и ногтей.
Консилиум врачей в лице представителей кафедры пропедевтики педиатрии № 2 и кафедры медицинской генетики, заведующей отделением патологии новорожденных, офтальмолога, лор-врача, дерматолога пришел к следующему заключению: клинические проявления заболевания — сухость, покраснение кожи, дистрофические изменения ногтей, алопеция, редкие брови и ресницы, дистрофические изменения роговицы, язвы роговицы, наличие признаков заболевания с первых дней жизни — дают основание диагностировать у ребенка синдром Сентера (КИД-синдром), относящийся к наследственным заболеваниям с аутосомно-доминантным типом наследования. У ребенка имеет место гипотрофия II степени. Диагноз экфолиативного дерматита снят, сепсис не подтвержден.
Ребенок продолжает находиться в Харьковской ОДКБ № 1.
Краткая литературная справка
Текстура кожи детерминирована генетически. Многие кожные заболевания, сопровождающиеся нарушением ороговения, являются врожденными; ихтиоз — один из симптомов ряда редких наследственных заболеваний. В литературе имеются сообщения о сочетании ихтиоза с патологией других органов [1–4].
Синдром Сентера (кератит — ихтиоз — глухота, КИД-синдром; keratitis, ichthyosis, deafness — KID) — редкая форма эктодермальной дисплазии, обусловленной мутациями в гене GIB2 (ген коннексина 26) с характерным поражением кожи, волос, органов зрения и слуха. Основными клиническими проявлениями являются сосудистый кератит, тяжелая сенсоневральная тугоухость и прогрессирующая кератодермия (номер в каталоге наследственных заболеваний OMIM 148210). Оба значения КИД впервые предложили B. Skinner с соавторами в 1981 г. для триады клинических признаков, которая указывала на нарушение в развитии и дифференцировке многослойного плоского эпителия.
Согласно результатам исследований, молекулярной причиной КИД-синдрома являются определенные мутации в гене GIB2. Мутации в гене коннексина 26 ответственны по крайней мере за 5 кожных заболеваний, при которых отмечается нарушение слуха. Заболевания эти совпадают по некоторым клиническим признакам, охватывая широкий спектр прявлений. Различия связаны лишь с тем, какие органы поражаются и какова тяжесть клинических симптомов [2, 5].
Исследования G. Rihard и соавторов доказали, что при КИД-синдроме с доминантным наследованием имеет место мутация гена, приводящая к замене аминокислоты в высококонсервативном первом внеклеточном домене анексинового белка. Замена гуанина на амин в 148-м положении последовательности ДНК приводит к изменению последовательности аминокислот, а именно к замене аспарагина на аспарагиновую кислоту в позиции 50. Предполагается, что данное изменение в консервативной области белка нарушает взаимодействие коннексонов соседних клеток и ограничивает транспорт ионов между клетками [6, 7].
Клиническая картина заболевания выражена с момента рождения, спектр кожных проявлений широк и включает эритродермию, сухость кожи с образованием тонких чешуек. Фотофобия, кератит и прогрессирующая неоваскуляризация роговицы описаны в 79 % случаев. Среди пациентов отмечены повышенная восприимчивость слизистых оболочек и кожи к инфекциям, а также сквамозно-клеточные карциномы [4, 8, 9]. В 10–23 % случаев возможно врожденное отсутствие волос, но как правило, волосы редкие, тонкие и тусклые. В единичных случаях при КИД-синдроме наблюдали лейконихию и узловатые пальцы. Последнее указывает на то, что клиника синдрома частично перекрывается проявлениями других заболеваний [6].
Известно, что в динамике наблюдения детей с КИД-синдромом отмечается сохранение характерного поражения кожи, наличие доречевой сенсоневральной тугоухости и поражения органов зрения вплоть до слепоты.
Современные молекулярно-генетические исследования могут позволить дифференцировать отдельные формы заболевания кожи, глаз, сенсоневральной тугоухости, однако вопросы их лечения и профилактики остаются нерешенными. Продолжение молекулярно-генетических исследований в совокупности с клиническими наблюдениями в дальнейшем должно открыть новые возможности для диагностики, терапии и профилактики этих заболеваний. На современном уровне наших диагностических технологий необходимо использование генетического консультирования с определением возможного риска заболевания, дородовой диагностики пока не существует. Изучается эффективность применения изотретиноина, доказаны побочные эффекты высоких доз этого препарата, рекомендуется лечение инфекций на фоне этого заболевания, совершенствование терапии расстройств слуха, оптимизация школьного образования для детей с данной патологией.
Список литературы
1. MSD, Мерк, Шарп и Доуш. Руководство по медицине. — М.: Мир. — Т. II. Нарушение кератинизации. — С. 624-625.
2. Маркова Т.Г. Диагностика синдрома кератина-ихтиоза-глухоты (КИД-синдром) / Т.Г. Маркова, Н.Б. Бражкина, А.В. Поляков и др. // Вестник отоларингологии. — 2012. — 3. — С. 58-61.
3. Zhang X. B. Mutation of GIB2 in a Chinese patient with keratitis-ichthyosis-deafness syndrome and brain malformation / X.B. Zhang, S.C. Wei, C.X. Hi et al. / Clin. Exp. Dermatol. — 2009. — 34. — 309-313.
4. Nygnist G.G. Malignant proliferating pillar fumors arising in KID syndrome: a report of two patient / G.G. Nygnist, C. Mumm, R. Gran et al. // Am. I. Med. Genet. — 2007. — 734-741.
5. Caceres-Rios H. Kerathyosis, and defness (KID sindrome), prevue of the literature and proposed of a new terminology / H. Caceres-Rios, Tamayo-Sanchez C., Duran-McKinser O.M. et al. // Pediatr. Dermatol. — 1996. — 13, 2. — 105-113.
6. Kone-Poul I., Hesse S., Palix C. et al. Keratitis, ichthyosis and deafness (KID syndrome) in half sibs // Pediat. Dermatol. — 1998. — 15, 3. — 219-221.
7. Richard G. Messene mutations in GIB2 encoding connexin-26 the ectodermal desplasia keratitis-ichthyosis-deafness syndrome / G. Richard, F. Rouan, C.E. Willaughby et al. // Am. I. Hum Ge-net. — 2002. — 70. — 1341-1348.
8. Alvares A.S. De novo mutation in the gene encoding connexin-26 (GIB2) in a sporadic case of keratitis- ichthyosis-deafness (KID) syndrome / A.S. Alvares, M. Pera, M.A. Villamar // Am. I. Med. Genet A. — 2003. — 117. — P. 89-91.
9. Nyguist G.G. Malignant proliferation pillar tumors arising in KID syndrome: a report of two patients / G.G. Nyguist, C. Mumm, R. Grau et al. // Am. I. Med. Genet. — 2007. — 143. — 734-741.
10. Griffith A.J. Cochleosaccular dysplasia associrted with a connexin 26 mutation in keratitis-ichthyosis-deafness syndrome / A.J. Griffith, I. Iong, S.P. Prior et. аl. // Laringoscope. — 2006. — 116. — 1404-1408.